 Solutions
SolutionsLa estrategia de control de la contaminación es un concepto clave que define la nueva versión del Anexo 1. La apuesta por la mayor automatización y digitalización de los procesos como catalizador de la mejora en continuo es un factor relevante para el aumento de la eficiencia de los procesos y la calidad del producto. Bajo este foco, disponer de los datos clave es un asunto esencial que debe afrontar cualquier fabricante, para aumentar los niveles de conocimiento de sus procesos, disminuir la incertidumbre, y facilitar así la toma de decisión basada en el riesgo.
Introducción a la Estrategia de control de la contaminación
El nuevo Anexo 1 sobre Fabricación de medicamentos estériles proporciona una guía detallada para la fabricación de medicamentos estériles y contiene requisitos específicos para garantizar la seguridad, la calidad y la eficacia de los productos estériles. La nueva guía cubre todos los aspectos de la fabricación, desde el diseño de las instalaciones y el control ambiental, hasta la formación del personal y las actividades de validación correspondientes.
La nueva versión hace foco en una mejor aplicación del proceso de gestión de los riesgos (ICH Q9 – Quality Risk Management) y del sistema de calidad farmacéutico (ICH Q10 – Pharmaceutical Quality System) para asegurar un mayor control de la contaminación. En este ámbito, el nuevo anexo enfatiza la implementación de una estrategia de control de la contaminación (CCS, Contamination Control Strategy, por sus siglas en inglés) para definir los puntos críticos de control y evaluar la eficacia de todos los controles (de diseño, procedimiento, técnicos y organizativos), así como las medidas de seguimiento empleadas para minimizar el riesgo de contaminación.
En consecuencia, las organizaciones deben realizar un ejercicio de revisión del grado de conocimiento y experiencia de sus procesos de fabricación (instalaciones y equipos de producción, servicios farmacéuticos, métodos y tecnologías aplicadas) y de su sistema de calidad implementado, con el fin de controlar los factores que pueden afectar a la calidad de sus productos.
A su vez, el nivel de esfuerzo y de detalle de la estrategia de control de la contaminación debe ser proporcional al tipo de proceso y producto, ya que el nuevo Anexo 1 no solo provee directriz sobre la fabricación de productos «estériles» sino que también brinda orientación sobre la fabricación de los productos “no estériles”.
Por otro lado, la estrategia de control de la contaminación debe revisarse activamente y, ser actualizada correspondientemente para impulsar la mejora continua de los métodos de fabricación y control. Su eficacia debe formar parte de la gestión de los procesos de revisión periódica.
Elementos de la estrategia de control de contaminación
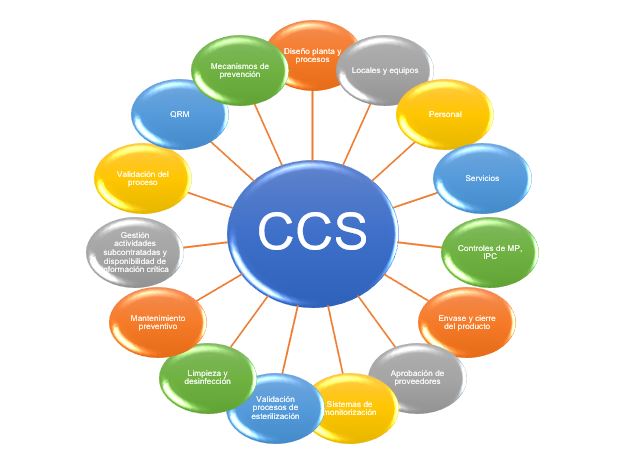
Tal y como menciona el Anexo 1, los elementos a considerar dentro de un CCS deben ser (pero no limitados a éstos) los siguientes:
- Diseño de la planta y de los procesos, incluyendo la documentación asociada.
- Locales y equipos.
- Personal.
- Servicios.
- Controles de materias primas, incluyendo los controles en proceso.
- Envases de productos y cierres.
- Aprobación de proveedores.
- Gestión de actividades subcontratadas y disponibilidad/transferencia de la información crítica entre las partes (ej.: servicios de esterilización).
- Proceso de gestión de riesgos.
- Validación del proceso.
- Validación de procesos de esterilización.
- Mantenimiento preventivo: mantenimiento de equipos, servicios e instalaciones (mantenimiento planificado y no planificado) para asegurar que no haya riesgo adicional de contaminación.
- Limpieza y desinfección.
- Sistemas de monitorización, incluyendo una evaluación de la viabilidad de la introducción de métodos alternativos científicamente sólidos que optimicen la detección de la contaminación ambiental.
- Mecanismos de prevención: análisis de tendencias, investigación detallada, determinación de la causa raíz, acciones correctivas y preventivas (CAPA) y la necesidad de herramientas de investigación exhaustivas.
- Mejora continua en base a la información derivada de lo anterior.
Figura 1: Elementos CCS.
¿Qué ofrece la digitalización?
El uso de tecnologías digitales avanzadas y de procesos de fabricación automatizados permite a las organizaciones disponer de mayores niveles de exactitud y precisión, al disminuir los niveles de incertidumbre, lo que se traduce en procesos más eficientes y en la fabricación de productos de mayor calidad. La monitorización en tiempo real y la mayor recopilación de datos permiten una mejor y más rápida detección de los problemas de calidad, así como del análisis de su causa raíz, lo que se traduce en un mejor control de la estrategia de control de contaminación, y del proceso de fabricación en general.
El cómo afrontar el proyecto de transformación digital, así como en seleccionar la información relevante, con los modelos adecuados para su tratamiento, suponen un desafío frente a la modernización de nuestros procesos. Es por ello imperativo, cuestionarse sobre la gestión de esta información, y si se hace de la forma óptima y eficiente.
Todo esto es en consecuencia, a la cada vez mayor complejidad de los sistemas informáticos, sus interconexiones, sumado al gran volumen y tipología de datos existente. A su vez, el gran potencial de nuevos sistemas inteligentes con disposición de análisis predictivos representa también un nuevo reto.
Nuestra toma de decisión debe estar basada en el riesgo para la protección del paciente, en arreglo al conocimiento científico de las etapas críticas y establecimiento de los sistemas, equipos y operaciones involucrados, así como de los datos de monitorización ambiental y del proceso resultantes.
Este punto es especialmente crucial, al abordar los sistemas de monitorización ambiental y de los procesos. Recordar, que el programa de monitorización ambiental y monitorización de procesos está compuesto por los siguientes elementos:
- Monitorización ambiental – partículas no viables.
- Monitorización ambiental y de personal – partículas viables.
- Temperatura, humedad relativa y otras características específicas.
- Simulación de procesos asépticos (sólo para productos de fabricación aséptica).
En esta andadura, es importante evaluar la ayuda que nos puede brindar en la visión holística de la estrategia de control de contaminación de nuestra instalación, el uso de nuevas tecnologías de fabricación estéril existentes (ej.: sistemas RABS, aisladores, sistemas robóticos), así como por ejemplo, el uso de contadores de partículas BFPCs en la monitorización en continuo ambiental de partículas viables, y cómo estos datos son correctamente integrados dentro de nuestro conjunto global de datos para el correcto seguimiento de nuestro CCS, y toma de decisión basada en el riesgo.
En todo este proceso, es por ello, un punto clave que las organizaciones afronten el análisis de sus datos, en donde la aplicación de las actividades de gestión del conocimiento, gestión de riesgos para la calidad (QRM) y el pensamiento crítico actúan como facilitadores del proyecto de transformación digital.
La implementación de nuevos sistemas de análisis de big data permiten analizar los datos estadísticos de nuestros procesos en modelos múltiples en lugar de un análisis de modelo único, para su mejor evaluación, y toma de decisión en la mejora de nuestra estrategia CCS.
La idea es disponer de sistemas inteligentes con análisis de datos proactivos y predictivos que nos ayuden a mirar hacia adelante, en lugar de hacia atrás (reactivo).
La digitalización puede ser un catalizador de la mejora continua ya que acelera nuestro nivel de conocimiento para la toma de decisión acertada.
Sistemas robotizados en la fabricación aséptica
El uso de sistemas robotizados son una solución ideal para proporcionar la operación altamente precisa y repetitiva que exige el proceso aséptico de forma segura. Además, los robots por su gran grado de flexibilidad y escalabilidad pueden operar en entornos donde para los humanos no es conveniente, siendo una característica especialmente relevante, ante operaciones de trabajo en áreas asépticas críticas, ya que generan niveles de partículas viables y no viables extremadamente bajos compatibles con los entornos ISO 5 más exigentes, y o pueden ser integrados en entornos operativos que requieren la contención de compuestos altamente activos.
Mirando al futuro
La transformación digital no es un cambio simple ni rápido. Los beneficios, sin embargo, son claros y las herramientas están disponibles. En este punto, la robótica y las nuevas tecnologías de fabricación estéril y de monitorización en continuo, junto con las herramientas de digitalización, que ofrecen mayor capacidad de reporte/análisis, son elementos clave para este nuevo reto y promover la mejora en continuo y la optimización de nuestra CCS.
Figura 2: Mejora continua de CCS
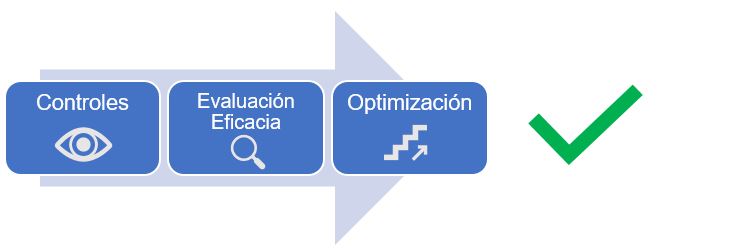
Las nuevas tecnologías deben ser afrontadas bajo un Sistema de Gestión de Calidad que promueva este cambio tecnológico, en donde el pensamiento crítico, la aplicación del QRM y el papel de los proveedores tecnológicos como expertos son elementos clave para así afrontar la aversión al riesgo, en donde muchas veces es la principal barrera a superar.
Trescal
Desde Trescal, ofrecemos servicios de consultoría de calidad especializados, así como de validación, cualificación y calibración. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar el correcto desarrollo y/o optimización de su CCS, así como de afrontar con éxito la consecución de su proyecto de transformación digital para la mejora de los procesos de fabricación estéril, monitorización en continuo, así como de reporte/análisis desde la órbita de la calidad, no dude en contactar con nosotros.
Acrónimos
[1] CCS: Contamination Control Strategy
[2] QRM: Quality Risk Management
[3] CAPA: Acciones correctivas y preventivas
[4] MP: Materia prima
[5] IPC: In-process Controls
[6] RABS: Restricted Access Barrier System
[7] BFPC: Bio-Fluorescent Particle Counters
 GMP Qualification and validation
GMP Qualification and validation
 Hands placing last piece of a Puzzle
Hands placing last piece of a Puzzle
 Solutions
Solutions
 Solutions
Solutions imagen-1080×595
imagen-1080×595