
Introducción
El nuevo anexo clarifica cómo las empresas fabricantes pueden aprovechar las nuevas posibilidades derivadas de la aplicación de una comprensión mejorada del proceso de gestión de los riesgos (ICH Q9 – Quality Risk Management) y del sistema de calidad farmacéutico (ICH Q10 – Pharmaceutical Quality System), así como incluye nuevos desarrollos tecnológicos y metodologías que han hecho necesaria la revisión del Anexo 1.
Además, es importante remarcar que el nuevo anexo reconoce en su alcance que aunque su intención es dar orientación en la fabricación de productos estériles, algunos de los principios y orientaciones, como la estrategia de control de la contaminación, el diseño de las instalaciones, la clasificación de salas limpias, la cualificación, la validación, el control y la vestimenta del personal, pueden utilizarse para respaldar la fabricación de otros productos no estériles, en donde el control y la reducción de la contaminación microbiana, de partículas y de endotoxinas/pirógenos se considera importante.
Estrategia de control de la contaminación y PQS
La CCS es uno de los ejes clave de la revisión del nuevo anexo 1 y uno de los que ha generado mayor controversia. Pero la CCS, no es ni mucho menos, un aspecto desconocido para las empresas fabricantes de productos farmacéuticos, ya que la CCS es un factor clave para garantizar la calidad del producto, y el óptimo desempeño de los procesos de fabricación.
Lo que ha ocurrido es que con la nueva revisión del anexo 1, se ha querido formalizar esta materia para brindar guía a la industria para la disposición de estrategias de control más efectivas, con la inclusión de los elementos clave a tener en cuenta, y dar alineación dentro del proceso de gestión de los riesgos y del sistema de calidad farmacéutico. De hecho, se trata de mejorar el sistema de calidad aplicado, para dotarlo de las correctas prácticas, y metodologías, con sistema de CAPA, desviaciones, No conformidades con análisis causa raíz, CC, gestión de la revisión, QRM integrado, monitorización y desempeño de los procesos y calidad del producto (QTPP – Quality Target Product Profile-), gestión del conocimiento, y con foco en su optimización y mejora continua.
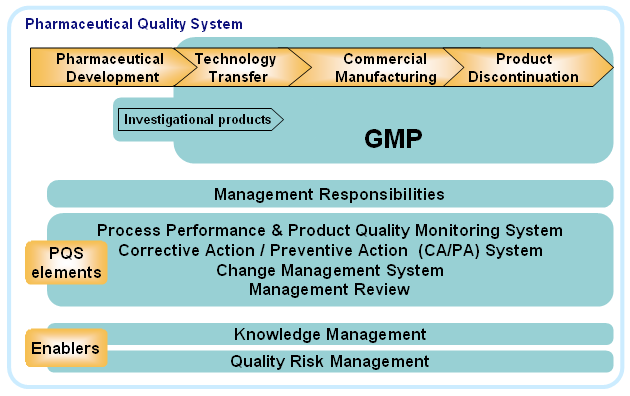
Imagen 1: Pharmaceutical Quality System. Fuente: ICH Q10. Annex 2.
El objetivo es conseguir un sistema maduro (Quality Managament Maturity), con menos defectos de calidad, y la consiguiente reducción de retiradas de producto, problemas de desabastecimiento, así como daños en la reputación de la compañía.
Elementos a evaluar
La nueva revisión del anexo 1 ya nos ofrece guía, en su capítulo 2.5 de Principios, sobre los elementos a considerar en orden de minimizar los riesgos por contaminación microbiológica, partículas y endotoxinas/pirógenos. Pero para implementar nuestra estrategia de control de contaminación (CCS) debemos revisar cada uno de los puntos recogidos a lo largo de todo el anexo 1, para la inclusión de los nuevos requerimientos, así como de las recomendaciones y requerimientos implícitos que dependen del resultado de un ejercicio de gestión de riesgos previo.
Los elementos propuestos a ser considerados para el CCS son (pero no limitados a éstos) los siguientes:
- Diseño de la planta y de los procesos, incluyendo la documentación asociada.
- Locales y equipos.
- Personal.
- Servicios.
- Controles de materias primas, incluyendo los controles en proceso.
- Envases de productos y cierres.
- Aprobación de proveedores.
- Gestión de actividades subcontratadas y disponibilidad/transferencia de la información crítica entre las partes (ej.: servicios de esterilización).
- Proceso de gestión de riesgos.
- Validación del proceso.
- Validación de procesos de esterilización.
- Mantenimiento preventivo: mantenimiento de equipos, servicios e instalaciones (mantenimiento planificado y no planificado) para asegurar que no haya riesgo adicional de contaminación.
- Limpieza y desinfección.
- Sistemas de monitorización, incluyendo una evaluación de la viabilidad de la introducción de métodos alternativos científicamente sólidos que optimicen la detección de la contaminación ambiental.
- Mecanismos de prevención: análisis de tendencias, investigación detallada, determinación de la causa raíz, acciones correctivas y preventivas (CAPA) y la necesidad de herramientas de investigación exhaustivas.
- Mejora continua en base a la información derivada de lo anterior.
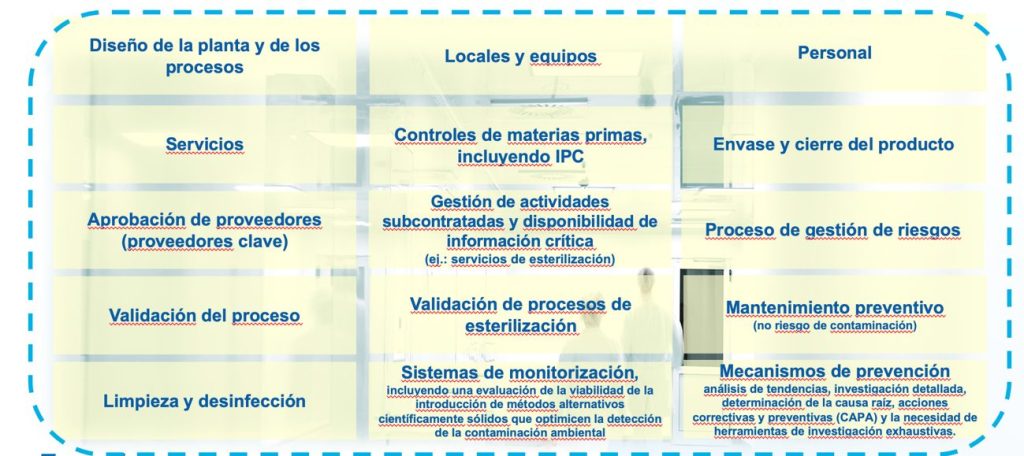
Imagen 2: Elementos a evaluar
Debemos conocer y comprender de dónde provienen las fuentes de contaminación potencial en las instalaciones y procesos, así como preguntarnos cómo podría ingresar y proliferar dicha contaminación, cuáles y cómo son nuestros controles y sistemas de monitorización actuales, y cómo deberían ser con el fin de buscar estrategias de control más efectivas para una mejor reducción y detección de los problemas de contaminación.
Conclusión
El nuevo anexo 1 es un buen recordatorio para la revisión y mejora de las estrategias de control de la contaminación dentro del PQS y uso de QRM. En consecuencia, debemos revisar nuestro sistema de gestión de Calidad, procesos de fabricación e instalaciones para su correcta alineación, contando con los expertos en la materia.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, validación de sistemas, cualificación de equipos y calibración. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten:
- Establecimiento de los requerimientos técnicos y reguladores.
- Revisión del diseño.
- Gap análisis de la estrategia actual de control de la contaminación.
- Evaluación de proveedores.
- Cualificación de salas limpias.
- Ensayos de direccionalidad de humo (visualización y filmación)
- Cualificación de servicios farmacéuticos.
- Cualificación de equipos de esterilización.
- Establecimiento y mantenimiento de los planes de verificación de salas limpias.
- Desarrollo / revisión de la estrategia de control de la contaminación (CCS).
- Validaciones de proceso.
- Validaciones de limpieza.
- Desarrollo de los expedientes de validación / cualificación.
- Acompañamiento a proveedores en pruebas de aceptación.
- Revisiones periódicas.
- Calibración instrumental.
- Actividades de formación dirigida.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar la correcta alineación de su sistema de calidad, procesos e instalaciones con los nuevos requisitos del Anexo 1, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.



