Tal y como vimos en el anterior artículo (Parte 1) el software de un dispositivo médico / producto sanitario debe ser implementado de una forma segura y efectiva para asegurar el uso y seguridad del producto final.
El estándar IEC 62304, de aplicación tanto para el software incorporado en un dispositivo, como para el software que es un dispositivo médico en sí mismo, sirve como referencia para todo el ciclo de vida del software de dispositivos médicos.
A continuación, echaremos un vistazo a las actividades de Verificación y Validación (V&V) dentro del proceso de desarrollo del software.
Verificación y validación
Las actividades de verificación y validación (V&V) son fundamentales para la obtención de dispositivos médicos seguros.
Mientras, la verificación se refiere a la comprobación del producto desarrollado conforme a sus requisitos especificados, la validación comprueba si se cumple su uso previsto en arreglo a las necesidades del usuario. De forma general, los términos de verificación y validación responden cada uno, a las siguientes preguntas:
- Verificación: ¿El diseño del dispositivo cumple los requisitos definidos?
- Validación: ¿Se cumplen los requisitos para el uso previsto del dispositivo? ¿Se ha diseñado el dispositivo adecuado?
A su vez, las actividades de verificación y validación en el proceso de diseño de dispositivos médicos / producto sanitario, deben estar respaldadas por un sólido plan de gestión de riesgos, en línea con las normas ISO 14971, y cumplir con los procedimientos del sistema de gestión de calidad correspondiente (alineado con ISO 13485).
Es importante tener en consideración que dentro del ejercicio de gestión de riesgos se haya identificado el diseño más crítico o el peor caso, considerando los riesgos identificados y los resultados. Puede ser necesario evaluar varios diseños críticos para demostrar un control efectivo de los riesgos asociados.
El nivel de formalidad de las pruebas será determinado a su vez bajo un ejercicio de gestión de riesgos.
A continuación, se muestra el diagrama en cascada de los controles de diseño de la FDA.
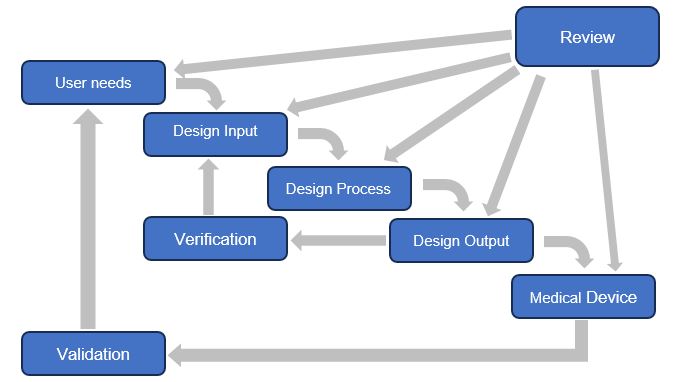
Figura 1: Aplicación de controles de diseño al proceso de diseño en cascada (figura utilizada con autorización de la Oficina de Dispositivos Médicos, Health Canada)
En resumen, el propósito principal de las actividades de verificación y validación de diseño es garantizar que el dispositivo se encuentra dentro de los límites del diseño especificado, cumplan con las necesidades del usuario y de su uso previsto, dentro de un proceso controlado y trazable.
Actividades de verificación
Las actividades de verificación se llevan a cabo en todas las etapas y niveles del diseño del dispositivo. Cualquier enfoque que establezca la conformidad con un requisito de entrada del diseño es un medio aceptable para verificar el diseño con respecto a ese requisito. En muchos casos, son posibles una variedad de enfoques. En línea con la IEC 62304, podemos distinguir diferentes niveles de verificación:
- Ensayos de la unidad del software. Ensayos dirigidos a verificar la implementación del diseño para cada elemento de software.
- Ensayos de integración del software. Ensayos que verifican la completa integración del elemento de software en el sistema software. Se trata de un proceso progresivo hasta la completa integración del sistema software. Incluye los ensayos de regresión que comprueban las capas adicionales de software conforme no introducen errores en el software previamente integrado.
- Ensayos del sistema software. Ensayos dirigidos a verificar el sistema de software de forma completa.
Actividades de Validación
Las actividades de validación del diseño prosiguen a las de verificación, una vez éstas han concluido de forma exitosa y ya se dispone del dispositivo acabado o equivalente. Esta actividad puede incluir unidades de producción inicial, lotes o sus equivalentes con la justificación de la elección del producto, y su evaluación clínica.
Los pasos a seguir en el proceso de validación del diseño son los siguientes:
- Desarrollo del Plan de validación. Se trata de un documento de sistemática, definición de alcances y estrategia. Este documento debe incluir la identificación del alcance de las actividades de validación, así como su enfoque de especificación/ensayo, restricciones posibles, criterios de aceptación, entornos de trabajo, recursos, cronología, y metodologías a aplicar. Es importante revisar la incorporación al plan del cumplimiento de los requisitos reguladores , así como hacer referencia a otro tipo de actividades relacionadas, como por ejemplo, las de especificación/verificación, o de evaluación clínica.
- Desarrollo de un ejercicio de evaluación de riesgos integrado y completo teniendo en cuenta el uso previsto, y los factores de influencia secundarios, como componentes de interfaz del sistema/producto final o el modo de montaje.
- Redacción del Plan de pruebas que incluya los criterios de aceptación y los resultados. Deben incluirse los casos de test con los escenarios comunes que demuestren que el software cumple con su uso previsto. Están diseñados para descubrir errores potenciales y demostrar si las funciones clave funcionan correctamente.
- Ejecución del Plan de pruebas en arreglo a los procedimientos de prueba, los requisitos reglamentarios, y buenas prácticas documentales.
- Informe final de validación con el resumen de resultados e información de soporte para la adecuada liberación del software. Es la respuesta al Plan de Validación. En esta etapa, se aprovecha a su vez para la revisión de los procedimientos de uso / instrucciones, así como el riesgo residual final.
Remarcar que las actividades de verificación y validación no se limitan a una actividad única, ya que debe asegurase que el sistema software es conforme tras los cambios de configuración o mejoras que puedan llegar a producirse a lo largo de su ciclo de vida.
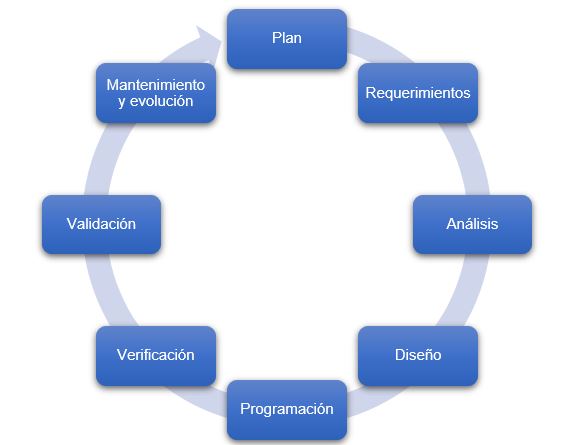
Figura 2: Ciclo de vida del software
Trazabilidad y automatización
Tanto para las actividades de Verificación, así como de validación, es muy importante contar con una Matriz de Trazabilidad de Requisitos (RTM) que asegure la correcta trazabilidad entre requisitos y ensayos de forma sencilla y eficaz.
A su vez, las nuevas herramientas existentes en el mercado para la automatización de las actividades a lo largo del desarrollo de software, así como de validación digital, hacen posible una mejor gestión de las actividades, permitiendo la trazabilidad de cada requisito, con su especificación, análisis, y ensayo/test de verificación y validación, incluyendo las evidencias, así como una reducción de los errores humanos.
Este enfoque para la mejora de la trazabilidad es especialmente útil para detectar suposiciones o premisas ocultas. Las suposiciones ocultas son peligrosas porque a menudo conducen a un diseño excesivo, lo que añade costes y complejidad innecesarios al diseño. En otros casos, resultan en un diseño no documentado.
Ciberseguridad
Las actuales regulaciones sobre dispositivos médicos están evolucionando para garantizar que los dispositivos comercializados sean aptos para los nuevos desafíos tecnológicos relacionados con los riesgos en materia de ciberseguridad.
En la actualidad, existen numerosos documentos guía publicados por los diferentes organismos reguladores que debemos tener en cuenta a la hora de implementar las medidas de ciberseguridad más adecuadas en nuestros dispositivos.
En especial debemos prestar atención a los requisitos esenciales de seguridad para todos los dispositivos médicos que incorporen sistemas electrónicos programables (PEMS, por sus siglas en inglés) y software que sean en sí mismos dispositivos médicos. Debemos aplicar en este campo principios de gestión de riesgos, incluida la seguridad de la información, así como establecer requisitos mínimos de seguridad informática, incluida la protección contra el acceso no autorizado.
Por otro lado, existen muchos dispositivos que no fueron diseñados con estas mismas consideraciones y exigencias, por lo que pueden presentar riesgos para los pacientes. Por ello, debemos abordar sus amenazas de ciberseguridad, tal y como recomiendan las regulaciones y buenas prácticas actuales.
Pensamiento crítico y enfoque CSA
El enfoque CSA de la CDRH (FDA) intenta clarificar la interpretación de los requisitos regulatorios con el entorno tecnológico actual, para cambiar el foco de la documentación al pensamiento crítico durante todo el ciclo de vida del software de un dispositivo médico.
La “garantía de la calidad del software” se debe centrar en prevenir la introducción de defectos en el proceso de desarrollo de software y fomenta el uso de un enfoque basado en el riesgo para establecer la confianza conforme el software es adecuado para su uso previsto.
Este nuevo enfoque promueve por lo tanto buenas prácticas de calidad orientadas en los siguientes conceptos:
- Aprovechar la experiencia de los expertos en la materia (SME) aplicando el pensamiento crítico para determinar la metodología y tecnología de verificación más apropiada basada en el riesgo.
- Generación de evidencias documentales de soporte siempre y cuando aporten un valor añadido a la calidad de las pruebas.
- Mayor dedicación a la verificación activa para la búsqueda de defectos y menor dedicación a la generación de especificaciones por adelantado.
- Cambio de foco desde la documentación al pensamiento crítico y a la verificación.
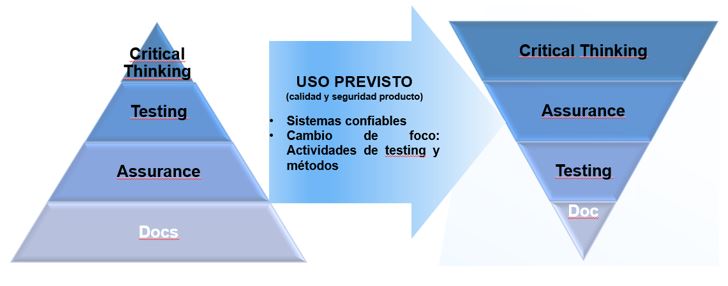
Figura 3: Enfoque CSA y pensamiento crítico
Machine Learning y Artificial Intelligence
En la actualidad se ha acelerado la adopción y el uso de tecnología AI/ML en dispositivos médicos. Este tipo de dispositivos tienen el potencial de transformar la atención médica al obtener conocimientos nuevos y relevantes en consecuencia de la gran cantidad de datos generados durante todas las fases del proceso de atención médica.
Uno de los mayores beneficios de este tipo de dispositivos reside en la oportunidad de seguir aprendiendo e iterando a medida que hay datos adicionales disponibles, incluso del uso y la experiencia en el mundo real, para mejorar su rendimiento.
Al ser aún una tecnología con una introducción aún incipiente, las entidades reguladoras están publicando documentos de reflexión y concepto sobre la materia para alertar a la industria sobre sus riesgos, para la no introducción de sesgos, clarificación de responsabilidades, mantenimiento de la privacidad, seguridad y confidencialidad de los datos, así como de no introducción de errores por una no adecuada fiabilidad y precisión de los resultados.
Normativas de referencia
Las empresas fabricantes de software de dispositivos médicos deben disponer de un sistema de gestión de calidad adecuado que asegure el correcto ciclo de vida del dispositivo médico. A continuación, se dan como referencia y orientación las siguientes guías y estándares:
Estándares
- ISO 13485 Medical Devices – Quality Management Systems – Requirements for Regulatory Purposes
- ISO 14971 Medical Devices – Application of Risk Management to Medical Devices
- ISO 14155 Clinical Investigation of Medical Devices for Human Subjects: Good Clinical Practice
- IEC 62366-1 Medical devices – Part 1: Application of usability engineering to medical devices
- IEC 62304:2006 Medical device software. Software life cycle processes.
- IEC 82304-1:2016 Health software – Part 1: General requirements for product safety
- ISO/TS 82304-2:2021 Health software – Part 2: Health and wellness apps. Quality and reliability
- IEC 81001-5-1 IEC 81001-5-1 Health software and health IT systems safety, effectiveness and security.
- ISO/IEC TR 24028:2020 Information technology — Artificial intelligence — Overview of trustworthiness in artificial intelligence
- ISO/IEC DIS 5259-1 Artificial intelligence — Data quality for analytics and machine learning (ML) Under development (draft 2023)
Documentos guías publicados por Autoridades Regulatorias
- CDRH. Draft Guidance – Computer Software Assurance for Production and Quality System Software. Sep 2022.
- Europe MDCG 2021-3, Questions and Answers on Custom-Made Devices (& considerations on Adaptable medical devices and Patient-matched medical devices). Mar 2021.
- US FDA 21 CFR 820.30, Design Control Guidance for Medical Device Manufacturers. Mar 1997.
- US FDA CDRH, Technical Considerations for Additive Manufactured Devices – Guidance for Industry and Food and Drug Administration Staff. Dec 2017.
- US FDA CDRH, Applying Human Factors and Usability Engineering to Medical Devices – Guidance for Industry and Food and Drug Administration Staff. Feb 2016.
- US FDA CDER, CBER – Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients – Guidance for Industry (Revision 1). Sept 2016.
- MITRE_MDIC (FDA) – Playbook for Threat Modeling Medical Devices.
- Cybersecurity for Networked Medical Devices Containing Off-the-Shelf (OTS) Software.
- MDCG 2019-16—Guidance on Cybersecurity for Medical Devices.
- Australia TGA, Guidance on Personalized Medical Devices (including 3D-printed Devices) regulatory reforms. 2022.
- Health Canada, Supporting Evidence for Implantable Medical Devices Manufactured by 3D Printing. Apr 2019.
- China NMPA, Technical Review Guidance for the Registration of Personalized Additive Manufacturing Medical Devices of Passive Implantable Bone, Joint and Oral Hard Tissues.
- Japan MHLW, Guidance on Evaluation of Customized Orthopedic Devices for Osteosynthesis. Dec 2010.
- Japan MHLW, Guidance on Evaluation of Orthopedic Customized Artificial Hip Joint Prosthesis. Dec 2011.
- Singapore HSA, Regulatory Guideline for 3D-Printed Medical Devices, July 2021
- South Korea MFDS, Guidance for Patient-matched Medical Devices manufactured using 3D printers. Dec 2015.
- Discussion Papers. AI in Drug Manufacturing, (March 2023);
- Using Artificial Intelligence and ML in the Development of Drug and Biological Products. FDA, CDER. May 2023.
- Reflection paper on the use of Artificial Intelligence (AI) in the medicinal product lifecycle. EMA, CVMP & CHMP. Jul 2023.
- GMLP for Medical Device Development: Guiding Principles. MHRA, Health Canada, FDA. Oct 2021.
Documentos guías publicados por IMDRF / GHTF
- GHTF/SG3/N99-10:2004 (Edition 2) Quality Management Systems – Process Validation Guidance.
- GHTF/SG1/N71:2012 Definitions of the Terms ‘Medical Device’ and ‘In Vitro Diagnostic (IVD) Medical Device’.
- GHTF/SC/N4:2012 (Edition 2) Glossary and definition of terms used in GHTF documents.
- IMDRF/ UDI WG/N48 FINAL: 2019 Unique Device Identification system (UDI) Application Guide.
- IMDRF/PMD WG/N49 Final: 2018 Definitions for Personalized Medical Devices.
- IMDRF/GRRP WG/N47 FINAL: 2018 Essential Principles of Safety and Performance of Medical Devices and IVD Medical Devices.
- IMDRF/MDCE WG/N55 FINAL: 2019 Clinical Evidence – Key Definitions and Concepts.
- IMDRF/MDCE WG/N56 FINAL: 2019 Clinical Evaluation.
- IMDRF/MDCE WG/N57 FINAL: 2019 Clinical Investigation.
- IMDRF/GRRP WG/N52 FINAL: 2019 Principles of Labelling for Medical Devices and IVD Medical Devices.
- IMDRF/PMD WG/N58 FINAL: 2023 Personalized Medical Devices – Regulatory Pathways.
- IMDRF/ MDCE WG/N65 FINAL: 2021 Post-Market Clinical Follow-Up Studies.
- IMDRF Principles and Practices for Medical Device Cybersecurity.
Documentos guías publicados por la industria
- ISPE. GAMP 5 Guide 2nd Edition. A Risk-Based Approach to Compliant GxP Computerized Systems. July 2022.
- ISPE. GAMP Good Practice Guide: Enabling Innovation. Sep 2021.
- ISPE. RDI GAMP Guides: Records and data integrity.
- AAMI TIR45:2023; Guidance on the use of agile practices in the development of medical device software.
¿Qué ofrece Trescal?
Desde TRESCAL ofrecemos servicios de soporte relativos al software de dispositivo médico según las normativas de aplicación, ofreciendo consultoría de calidad específica en las siguientes materias:
- Auditoría de software.
- Establecimiento de los requerimientos técnicos y reguladores.
- Revisión del diseño.
- Proceso de desarrollo y mantenimiento del software de dispositivo médico (IEC 62304).
- Desarrollo del VMP / VP.
- Establecimiento de estrategias.
- Desarrollo de los expedientes de validación.
- Actividades de verificación y validación.
- Transición a enfoque CSA (Computer Software Assurance).
- Cualificacion de Infraestructura IT
- Evaluación de proveedores de SW / tecnológicos.
- Acompañamiento a proveedores en pruebas de aceptación.
- Procedimientos del sistema de gestión de calidad.
- Actividades de formación dirigida.
TRESCAL es un proveedor global de servicios para la industria LIfe Sciences que ofrece servicios de implantación/consultoría en sistemas de calidad de productos sanitarios, gestión de riesgos, Data Integrity, validación de sistemas, cualificación de equipos, calibraciones (incluyendo calibraciones dentro del sector médico/sanitario). Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del proyecto de validación, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Acrónimos
AI: Artificial Intelligence
CDRH: Center for Devices and Radiological Health
CSA: Computer Software Assurance
FDA: Food and Drug Administration
GHTF: Global Harmonization Task Force
IMDRF: international Medical Device Regulators Forum
ML: Machine Learning
PMS: Programmable Electrical Medical Systems
RTM: Requirements Traceability Matrix
SME: Subject-Matter Expert
SW: Software



