TRESCAL PARTICIPARÁ EN FARMAFORUM LOS DÍAS 25 y 26 DE SEPTIEMBRE DE 2024
Madrid, 2 de septiembre, 2024. El grupo Trescal estará presente en Farmaforum la feria de referencia del sector farmacéutico este mes de septiembre.
Con el objetivo de seguir ampliando mercado e impulsar la imagen Trescal en el sector life sciences Trescal participará con el stand E32 en Farmaforum, Madrid. La feria tendrá lugar los días 25 y 26 de septiembre en IFEMA, Pabellón 9.
Un año más una oportunidad única para mantenerse al día sobre las últimas actualidades GxP y de los servicios más actuales.
A continuación, detallamos los talleres que ofrecerán:
25 de septiembre de 10 a 12h: Novedades del Anexo I
Ponente: Mar Díaz, Responsable Técnica Validaciones y Roberto Español, Responsable Técnico Trescal Life Sciences
¡Estaremos esperándote en nuestro stand! Puede conseguir invitaciones en el siguiente enlace
TECHNICAL_BNL_ANVERS_2021_PRESSURE (3)
La UPC y el grupo TRESCAL firman un acuerdo para fomentar las vocaciones STEAM entre los jóvenes
La UPC y el grupo TRESCAL, han firmado un convenio de colaboración a través del cual se apoyará a la organización, la gestión y la difusión del programa de Fomento de Vocaciones y Divulgación STEAM de la Universidad, dirigido en los centros educativos de Cataluña.
El convenio de colaboración entre grupo TRESCAL, y la Universitat Politècnica de Catalunya – BarcelonaTech (UPC) se materializará en una aportación de 5.000 euros por parte de la empresa destinados al programa de fomento de vocaciones STEAM de la UPC, a través del cual la Universidad ofrece un catálogo de actividades de fomento de vocaciones STEAM, dirigido principalmente a alumnado de primaria, ESO y bachillerato de centros educativos de Cataluña, así como a familias y a la sociedad en general.
Las actividades que organiza la UPC están orientadas a despertar vocaciones en los estudios STEAM (por sus siglas en inglés de ciencias, tecnología, ingeniería, artes –que incluye arquitectura, urbanismo y edificación– y matemáticas). El programa de actividades está orientado a romper los estereotipos de género y los asociados a las profesiones STEAM, explicar la aplicabilidad social de estas profesiones y dar visibilidad a la actividad de los investigadores y las investigadoras de la Universidad que llevan a cabo estas actividades.
Por su parte, el grupo TRESCAL tiene interés en patrocinar acciones en el ámbito de la educación universitaria con el fin de promover y apoyar acciones y actividades de fomento de vocaciones en ámbitos de especialización de la compañía en el marco de su programa de responsabilidad social corporativa.
La UPC tiene una amplia trayectoria en el desarrollo de proyectos y actividades orientados al fomento de las vocaciones STEAM a través de un amplio programa de actividades dirigido a los centros educativos, al alumnado de primaria, ESO y bachillerato, a sus familias y a la sociedad en general. La UPC ofrece a los centros educativos un amplio catálogo de conferencias de divulgación científica y tecnológica y talleres STEAM, que llevan a cabo personal docente e investigador de la Universidad. El último curso académico se han impartido 450 conferencias, a las que han asistido más de 15.700 jóvenes, y se han organizado 160 talleres STEAM, que han contado con la participación de más de 3.150 alumnos.
LinkedIn (1)
Trescal participará en ADM Sevilla, 14-16 Mayo
Tenemos el placer de anunciarles que el grupo Trescal estará presente en ADM en Sevilla los próximos días 14 – 16 de mayo 2024, la feria de referencia del sector aeroespacial y defensa en España. Con el objetivo de seguir ampliando mercado e impulsar la imagen Trescal en el sector aeroespacial y defensa Trescal participará con el stand D11 en ADM Sevilla en Sevilla City Office (antiguo FIBES Palacio de Congresos).
Nuevo marco de calidad de datos de la EMA / HMA para la regulación de medicamentos.
En diciembre de 2023, la EMA y la HMA publicaron conjuntamente la primera versión completa del documento sobre Marco de Calidad de Datos para la regulación de medicamentos en la EU. Este documento tiene como objetivo caracterizar, evaluar y garantizar la calidad de datos para el respaldo de la toma de decisiones regulatorias.
Introducción y foco
El progreso en la digitalización y la tecnología de la información ha creado nuevas oportunidades que han contribuido a un panorama cada vez más complejo para la toma de decisiones regulatorias. Por ello, se está reconociendo una creciente necesidad de estandarización y la implementación de un marco que describa la calidad de datos. Este marco permitiría al regulador realizar evaluaciones regulatorias confiables sobre si los datos son apropiados para respaldar la toma de decisiones. Pero, aunque se dispone de nuevos tipos o estándares de datos, todavía son escasas las directrices o métodos para demostrar si dichos datos son adecuados para la toma de decisiones.
Por todo ello, se ha visto necesario por parte de la European Medicines Agency (EMA) y la Heads of Medicines Agencies (HMA) la elaboración de un documento Marco de Calidad de Datos (DQF, por sus siglas en inglés) que brinde orientación y guía para el logro de procedimientos de evaluación de calidad coherentes y consistentes. Este documento de 42 páginas, ha sido publicado en el mes de diciembre de 2023, tras ser actualizado tras los comentarios recibidos durante su versión preliminar de octubre de 2022.
Con el establecimiento de un marco de calidad de datos la EMA y la HMA pretenden:
Mejorar la coherencia en la evaluación de la calidad de datos utilizados por los reguladores.
Facilitar el desarrollo de un enfoque estandarizado para la calidad de datos en todas las fuentes de datos.
Favorecer un uso más sistemático de los datos en la toma de decisiones regulatorias.
Apoyar la confianza de los interesados en los datos que respaldan las decisiones regulatorias.
Este marco va alineado además con el objetivo principal del plan de trabajo del Grupo Directivo Conjunto de la HMA-EMA sobre Big Data que es el de desarrollar pautas para el logro de un proceso fortalecido para la cualificación de datos a través de asesoramiento científico, y promover en los Estados miembros la adopción de registros electrónicos de salud, registros, datos genómicos y la disponibilidad segura de los datos.
Alcance y contenido
El documento es la primera publicación del Marco de Calidad de Datos de la EU (DQF) para la regulación de medicamentos y define principios y procedimientos de alto nivel que se aplican en todo el mandato regulatorio de la EMA. Por ello, brinda un análisis sobre las acciones y métricas de calidad de datos que deben considerarse en diferentes casos de uso e introduce un modelo de madurez para guiar la evolución de la automatización para respaldar la toma de decisiones regulatorias basada en datos.
El objetivo es proporcionar un conjunto de definiciones, principios y directrices para su aplicación de forma coherente en cualquier fuente de datos con el fin de caracterizar, evaluar y asegurar la calidad del conocimiento para la toma de decisiones regulatorias. Este marco está destinado además a abarcar el uso primario y secundario de los datos, así como metadatos e información de respaldo.
Sobre las verificaciones de calidad de datos, estas ocurren en varios pasos a lo largo del proceso de generación de evidencia y pueden incluir ciclos de retroalimentación iterativos tal y como se indica la línea discontinua de la figura 1.
Figura 1: Flujo de trabajo típico de procesamiento de datos en el proceso de generación de evidencia.
Por otra parte, dentro del alcance, el documento quiere hacer reseña del concepto de dato, que define como “cualquier activo de información que representa mediciones u observaciones y que pueden usarse para apoyar la toma de decisiones, directa o indirectamente a través del análisis”, así como del de Calidad de datos que define como “la idoneidad para el propósito de las necesidades de los usuarios en relación con la investigación en salud, la formulación de políticas y la regulación, y que los datos reflejan la realidad que intentan representar.”
Se remarca por lo tanto que la calidad de datos debe ser evaluada desde el punto de vista de su idoneidad para satisfacer las necesidades de los usuarios.
Por otra parte, el documento está dirigido principalmente a la red reguladora de medicamentos de la EU, pero la relevancia del contenido puede ser de interés para una gama más amplia de partes interesadas, como titulares de autorizaciones de comercialización, titulares de fuentes de datos, investigadores y asociaciones de pacientes.
Consideraciones en la implementación del DQF
Por otra parte, el nuevo documento marco proporciona un conjunto de observaciones y recomendaciones guía en la implementación del DQF para el logro de mayores niveles de madurez. El documento destaca las siguientes:
Asegurar la calidad en origen: Al diseñar procesos de recopilación y generación de datos, los aspectos que afectan a la calidad de datos deben abordarse de forma inicial ya que cuanto más se alejan los datos del contexto original, más difícil resulta corregir los problemas. Este asunto es particularmente relevante en el caso de los metadatos, ya que el conocimiento del contexto de generación de datos está presente solo en el momento de la generación.
Disponibilidad de datos maestros (MDM) y datos de referencia: La disponibilidad de MDM y datos de referencia tiene un impacto directo en la calidad de datos. A menudo es un requisito previo para la coherencia de los datos e incluso puede afectar la confiabilidad en algunos escenarios de producción de datos, ya que la información desconectada puede generar información errónea.
Alineación del SGC: Un Sistema de Gestión de Calidad es un enfoque formalizado adoptado por una organización que documenta procesos, procedimientos y responsabilidades para lograr políticas y objetivos de calidad (por ejemplo, Good Clinical Practices [GCP], Good Laboratory Practices [GLP] or Good Manufacturing Practice [GMP]). A su vez, estándares como la familia ISO 9000 definen el SGC en todas las industrias, pero se han desarrollado SGC más específicos para industrias o productos específicos. Dentro del sector Life Sciences podemos resaltar los siguientes estándares:
ISO 14155 y Directiva de la UE 2001/20/EC para GCP (datos de ensayos clínicos)
Quality System Regulation (QSR) e ISO 13485 para dispositivos médicos.
Directiva de la UE 2004/9/EC y 2004/10/EC para GLP (datos de investigación de laboratorio).
ISO 15189 e ISO 17025 (datos de laboratorios clínicos).
Controles sobre los sistemas informáticos: Ante el actual marco de digitalización, los sistemas informáticos utilizados para crear, modificar, mantener, archivar, recuperar o transmitir datos son un elemento crucial a tener en cuenta cuando hablamos de calidad de datos. Debemos por ello, asegurar que disponemos de sistemas informáticos efectivos, confiables y de alta calidad. La validación del sistema informático garantiza que el software se implementa adecuadamente y que existan los controles de proceso necesarios para usarlo de acuerdo a especificaciones, incluyendo la documentación asociada, el control de acceso, la gestión de proveedores y las auditorías. Por otro lado, debemos contar con un ciclo de vida de desarrollo de software que incluya un sistema de garantía de calidad del software para garantizar su diseño, desarrollo y verificación cuando el dato del SW es el propio producto (ej.: software de dispositivo médico).
El papel de los estándares ISO de la industria: La Organización Internacional de Normalización (ISO) ha elaborado normas que proporcionan marcos para la implementación de diversos aspectos de la gestión de datos, que se prueban en el campo y para las cuales se establecen plataformas, servicios de apoyo y organismos de certificación. Estos estándares a menudo se desarrollan para la implementación de industrias donde no se aplica la toma de decisiones regulatorias de la EMA.
ISO 9000: Describe los estándares para los sistemas de gestión de calidad en todos los niveles de una organización. Se podría considerar la adopción de esta norma si no se aplica ningún SGC específico de la industria.
ISO 8000: Describe los estándares de gestión de calidad de datos de una organización sometidos a un ciclo de mejora continua. La ISO 8000-150 especifica los principios fundamentales de la gestión de la calidad de datos maestros, centrada en los procesos, y los requisitos para la implementación, el intercambio de datos y la procedencia.
ISO 25012: Define un modelo general de calidad de datos para datos retenidos en un formato estructurado dentro de un sistema informático, típico de los datos considerados para la toma de decisiones regulatorias. Proporciona un marco para establecer requisitos de calidad de datos, medidas de calidad de datos y un plan para realizar evaluaciones de calidad de datos. En un modelo de Calidad de Datos se establecen las 15 características de Calidad de Datos que se deben tener en cuenta a la hora de evaluar las propiedades de un producto de datos determinado (exactitud, completitud, consistencia, credibilidad, actualidad, accesibilidad, conformidad, confidencialidad, eficiencia, precisión, trazabilidad, comprensibilidad, disponibilidad, portabilidad y recuperabilidad).
ISO 13485: Es la norma que especifica los requisitos de un sistema de gestión de la calidad para que una organización diseñe, construya y obtenga autorización para dispositivos médicos que cumplan sistemáticamente con los requisitos reglamentarios y del cliente.
Consideraciones sobre integridad de datos: ALCOA ++ es un marco para la integridad de los datos utilizado en toda la industria farmacéutica. Postula un conjunto de atributos que deben cumplir los datos y su documentación para que éstos sean Atribuibles, Legibles, Contemporáneos, Originales, y Acertados (ALCOA). El ++ se refiere a los siguientes atributos: Completo, Consistente, Duradero, Disponible, Trazable.Siguiendo esta premisa, debemos centrarnos en los determinantes fundamentales que afectan principalmente las dimensiones de Confiabilidad de los datos, pero también en las de Extensión, Coherencia y Oportunidad.
Figura 2: Atributos ALCOA ++
ATRIBUIBLE: Atribuible a la persona o sistema que genera los datos. Vinculado a la fuente de datos LEGIBLE: Fácilmente legible por ser humano (reúne toda la información para que dato sea completo) CONTEMPORÁNEO: Registrados en el momento de su adquisición ORIGINAL: Primer registro del dato, o “True Copie” que preserve el contenido y significado ACERTADO: Verificado como correcto. Sin error. Verificación de cálculos, algoritmos++ completo, consistente, duradero, disponible, trazable.
Controles sobre la Calidad de datos: Existen diferentes implementaciones posibles de controles de calidad de datos dependiendo si los hechos reales que representan los datos son conocidos y accesibles o no. En caso que los hechos reales sean conocidos, los datos se pueden probar mediante validación (versus los registros fuente que contienen estos hechos), aunque la validación puede ser costosa y consumir mucho tiempo, y a menudo requiere el uso de evaluadores si los hechos no están disponibles en forma estructurada y legible. Alternativamente, los datos se pueden probar mediante métricas de plausibilidad intrínsecas, evaluando el conjunto de datos con respecto a: otros datos del mismo conjunto de datos, diferentes rangos de referencia externos o otras tendencias plausibles (ciertos datos pueden ser válidos cuando se observan individualmente, pero la tendencia colectiva de todos los datos de un tipo debe seguir las distribuciones o tendencias esperadas).
Conclusiones
La calidad de datos es un elemento crucial para realizar todo el potencial de la regulación basada en datos y respalda la confianza de los pacientes y profesionales de la salud. El proyecto de Marco de Calidad de Datos para la regulación de medicamentos en la Unión Europea establece los criterios de calidad para los datos para garantizar que éstos sean aptos para el propósito de respaldar las decisiones de beneficio-riesgo.
Normativas de referencia
European Medicines Regulatory Network Data Standardisation Strategy. December 16th, 2021.
Big Data Steering Group, Big Data Workplan 2022-2025.
Good Practice Guide for the use of the Metadata Catalogue of Real-World Data Sources, EMA/787647/2022.
Guideline on computerised systems and electronic data in clinical trials. EMA/INS/GCP/112288/2023.
PIC/S Guidance. Good practices for data management and integrity in regulated GMP/GDP environments. PI 041-1. 1 July 2021.
Good Practice Guide for the use of the Metadata. Catalogue of Real-World Data Sources. EMA/787647/2022. V 1.0 (draft)
Acrónimos
DQF: Data Quality Framework
EMA: European Medicines Agency
HMA: Heads of Medicines Agencies
EU: European Union
MDM: Master Data Management
SGC: Sistema de Gestión de Calidad
ISO: International Organization for Standardization
EHDS: European Health Data Space
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Sciences que ofrece servicios de implantación/consultoría en sistemas de calidad, gestión de riesgos, Data Integrity, Data Governance, validación de sistemas informáticos, cualificación de equipos, calibraciones. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para adecuar su sistema de gestión de calidad al nuevo marco de calidad de datos, revisando estrategias y controles, o si necesita soporte en la realización de verificaciones y validaciones no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Imagen1-1
SW de dispositivos médicos / producto sanitario: ciclo de vida y validación. Parte 2
Tal y como vimos en el anterior artículo (Parte 1) el software de un dispositivo médico / producto sanitario debe ser implementado de una forma segura y efectiva para asegurar el uso y seguridad del producto final.
El estándar IEC 62304, de aplicación tanto para el software incorporado en un dispositivo, como para el software que es un dispositivo médico en sí mismo, sirve como referencia para todo el ciclo de vida del software de dispositivos médicos.
A continuación, echaremos un vistazo a las actividades de Verificación y Validación(V&V) dentro del proceso de desarrollo del software.
Verificación y validación
Las actividades de verificación y validación (V&V) son fundamentales para la obtención de dispositivos médicos seguros.
Mientras, la verificación se refiere a la comprobación del producto desarrollado conforme a sus requisitos especificados, la validación comprueba si se cumple su uso previsto en arreglo a las necesidades del usuario. De forma general, los términos de verificación y validación responden cada uno, a las siguientes preguntas:
Verificación: ¿El diseño del dispositivo cumple los requisitos definidos?
Validación: ¿Se cumplen los requisitos para el uso previsto del dispositivo? ¿Se ha diseñado el dispositivo adecuado?
A su vez, las actividades de verificación y validación en el proceso de diseño de dispositivos médicos / producto sanitario, deben estar respaldadas por un sólido plan de gestión de riesgos, en línea con las normas ISO 14971, y cumplir con los procedimientos del sistema de gestión de calidad correspondiente (alineado con ISO 13485).
Es importante tener en consideración que dentro del ejercicio de gestión de riesgos se haya identificado el diseño más crítico o el peor caso, considerando los riesgos identificados y los resultados. Puede ser necesario evaluar varios diseños críticos para demostrar un control efectivo de los riesgos asociados.
El nivel de formalidad de las pruebas será determinado a su vez bajo un ejercicio de gestión de riesgos.
Figura 1: Aplicación de controles de diseño al proceso de diseño en cascada (figura utilizada con autorización de la Oficina de Dispositivos Médicos, Health Canada)
En resumen, el propósito principal de las actividades de verificación y validación de diseño es garantizar que el dispositivo se encuentra dentro de los límites del diseño especificado, cumplan con las necesidades del usuario y de su uso previsto, dentro de un proceso controlado y trazable.
Actividades de verificación
Las actividades de verificación se llevan a cabo en todas las etapas y niveles del diseño del dispositivo. Cualquier enfoque que establezca la conformidad con un requisito de entrada del diseño es un medio aceptable para verificar el diseño con respecto a ese requisito. En muchos casos, son posibles una variedad de enfoques. En línea con la IEC 62304, podemos distinguir diferentes niveles de verificación:
Ensayos de la unidad del software. Ensayos dirigidos a verificar la implementación del diseño para cada elemento de software.
Ensayos de integración del software. Ensayos que verifican la completa integración del elemento de software en el sistema software. Se trata de un proceso progresivo hasta la completa integración del sistema software. Incluye los ensayos de regresión que comprueban las capas adicionales de software conforme no introducen errores en el software previamente integrado.
Ensayos del sistema software. Ensayos dirigidos a verificar el sistema de software de forma completa.
Actividades de Validación
Las actividades de validación del diseño prosiguen a las de verificación, una vez éstas han concluido de forma exitosa y ya se dispone del dispositivo acabado o equivalente. Esta actividad puede incluir unidades de producción inicial, lotes o sus equivalentes con la justificación de la elección del producto, y su evaluación clínica.
Los pasos a seguir en el proceso de validación del diseño son los siguientes:
Desarrollo del Plan de validación. Se trata de un documento de sistemática, definición de alcances y estrategia. Este documento debe incluir la identificación del alcance de las actividades de validación, así como su enfoque de especificación/ensayo, restricciones posibles, criterios de aceptación, entornos de trabajo, recursos, cronología, y metodologías a aplicar. Es importante revisar la incorporación al plan del cumplimiento de los requisitos reguladores , así como hacer referencia a otro tipo de actividades relacionadas, como por ejemplo, las de especificación/verificación, o de evaluación clínica.
Desarrollo de un ejercicio de evaluación de riesgos integrado y completo teniendo en cuenta el uso previsto, y los factores de influencia secundarios, como componentes de interfaz del sistema/producto final o el modo de montaje.
Redacción del Plan de pruebas que incluya los criterios de aceptación y los resultados. Deben incluirse los casos de test con los escenarios comunes que demuestren que el software cumple con su uso previsto. Están diseñados para descubrir errores potenciales y demostrar si las funciones clave funcionan correctamente.
Ejecución del Plan de pruebas en arreglo a los procedimientos de prueba, los requisitos reglamentarios, y buenas prácticas documentales.
Informe final de validación con el resumen de resultados e información de soporte para la adecuada liberación del software. Es la respuesta al Plan de Validación. En esta etapa, se aprovecha a su vez para la revisión de los procedimientos de uso / instrucciones, así como el riesgo residual final.
Remarcar que las actividades de verificación y validación no se limitan a una actividad única, ya que debe asegurase que el sistema software es conforme tras los cambios de configuración o mejoras que puedan llegar a producirse a lo largo de su ciclo de vida.
Figura 2: Ciclo de vida del software
Trazabilidad y automatización
Tanto para las actividades de Verificación, así como de validación, es muy importante contar con una Matriz de Trazabilidad de Requisitos (RTM) que asegure la correcta trazabilidad entre requisitos y ensayos de forma sencilla y eficaz.
A su vez, las nuevas herramientas existentes en el mercado para la automatización de las actividades a lo largo del desarrollo de software, así como de validación digital, hacen posible una mejor gestión de las actividades, permitiendo la trazabilidad de cada requisito, con su especificación, análisis, y ensayo/test de verificación y validación, incluyendo las evidencias, así como una reducción de los errores humanos.
Este enfoque para la mejora de la trazabilidad es especialmente útil para detectar suposiciones o premisas ocultas. Las suposiciones ocultas son peligrosas porque a menudo conducen a un diseño excesivo, lo que añade costes y complejidad innecesarios al diseño. En otros casos, resultan en un diseño no documentado.
Ciberseguridad
Las actuales regulaciones sobre dispositivos médicos están evolucionando para garantizar que los dispositivos comercializados sean aptos para los nuevos desafíos tecnológicos relacionados con los riesgos en materia de ciberseguridad.
En la actualidad, existen numerosos documentos guía publicados por los diferentes organismos reguladores que debemos tener en cuenta a la hora de implementar las medidas de ciberseguridad más adecuadas en nuestros dispositivos.
En especial debemos prestar atención a los requisitos esenciales de seguridad para todos los dispositivos médicos que incorporen sistemas electrónicos programables (PEMS, por sus siglas en inglés) y software que sean en sí mismos dispositivos médicos. Debemos aplicar en este campo principios de gestión de riesgos, incluida la seguridad de la información, así como establecer requisitos mínimos de seguridad informática, incluida la protección contra el acceso no autorizado.
Por otro lado, existen muchos dispositivos que no fueron diseñados con estas mismas consideraciones y exigencias, por lo que pueden presentar riesgos para los pacientes. Por ello, debemos abordar sus amenazas de ciberseguridad, tal y como recomiendan las regulaciones y buenas prácticas actuales.
Pensamiento crítico y enfoque CSA
El enfoque CSA de la CDRH (FDA) intenta clarificar la interpretación de los requisitos regulatorios con el entorno tecnológico actual, para cambiar el foco de la documentación al pensamiento crítico durante todo el ciclo de vida del software de un dispositivo médico.
La “garantía de la calidad del software” se debe centrar en prevenir la introducción de defectos en el proceso de desarrollo de software y fomenta el uso de un enfoque basado en el riesgo para establecer la confianza conforme el software es adecuado para su uso previsto.
Este nuevo enfoque promueve por lo tanto buenas prácticas de calidad orientadas en los siguientes conceptos:
Aprovechar la experiencia de los expertos en la materia (SME) aplicando el pensamiento crítico para determinar la metodología y tecnología de verificación más apropiada basada en el riesgo.
Generación de evidencias documentales de soporte siempre y cuando aporten un valor añadido a la calidad de las pruebas.
Mayor dedicación a la verificación activa para la búsqueda de defectos y menor dedicación a la generación de especificaciones por adelantado.
Cambio de foco desde la documentación al pensamiento crítico y a la verificación.
Figura 3: Enfoque CSA y pensamiento crítico
Machine Learning y Artificial Intelligence
En la actualidad se ha acelerado la adopción y el uso de tecnología AI/ML en dispositivos médicos. Este tipo de dispositivos tienen el potencial de transformar la atención médica al obtener conocimientos nuevos y relevantes en consecuencia de la gran cantidad de datos generados durante todas las fases del proceso de atención médica.
Uno de los mayores beneficios de este tipo de dispositivos reside en la oportunidad de seguir aprendiendo e iterando a medida que hay datos adicionales disponibles, incluso del uso y la experiencia en el mundo real, para mejorar su rendimiento.
Al ser aún una tecnología con una introducción aún incipiente, las entidades reguladoras están publicando documentos de reflexión y concepto sobre la materia para alertar a la industria sobre sus riesgos, para la no introducción de sesgos, clarificación de responsabilidades, mantenimiento de la privacidad, seguridad y confidencialidad de los datos, así como de no introducción de errores por una no adecuada fiabilidad y precisión de los resultados.
Normativas de referencia
Las empresas fabricantes de software de dispositivos médicos deben disponer de un sistema de gestión de calidad adecuado que asegure el correcto ciclo de vida del dispositivo médico. A continuación, se dan como referencia y orientación las siguientes guías y estándares:
Estándares
ISO 13485 Medical Devices – Quality Management Systems – Requirements for Regulatory Purposes
ISO 14971 Medical Devices – Application of Risk Management to Medical Devices
ISO 14155 Clinical Investigation of Medical Devices for Human Subjects: Good Clinical Practice
IEC 62366-1 Medical devices – Part 1: Application of usability engineering to medical devices
IEC 62304:2006 Medical device software. Software life cycle processes.
IEC 82304-1:2016 Health software – Part 1: General requirements for product safety
ISO/TS 82304-2:2021 Health software – Part 2: Health and wellness apps. Quality and reliability
IEC 81001-5-1 IEC 81001-5-1 Health software and health IT systems safety, effectiveness and security.
ISO/IEC TR 24028:2020 Information technology — Artificial intelligence — Overview of trustworthiness in artificial intelligence
ISO/IEC DIS 5259-1 Artificial intelligence — Data quality for analytics and machine learning (ML) Under development (draft 2023)
Documentos guías publicados por Autoridades Regulatorias
CDRH. Draft Guidance – Computer Software Assurance for Production and Quality System Software. Sep 2022.
Europe MDCG 2021-3, Questions and Answers on Custom-Made Devices (& considerations on Adaptable medical devices and Patient-matched medical devices). Mar 2021.
US FDA 21 CFR 820.30, Design Control Guidance for Medical Device Manufacturers. Mar 1997.
US FDA CDRH, Technical Considerations for Additive Manufactured Devices – Guidance for Industry and Food and Drug Administration Staff. Dec 2017.
US FDA CDRH, Applying Human Factors and Usability Engineering to Medical Devices – Guidance for Industry and Food and Drug Administration Staff. Feb 2016.
US FDA CDER, CBER – Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients – Guidance for Industry (Revision 1). Sept 2016.
MITRE_MDIC (FDA) – Playbook for Threat Modeling Medical Devices.
Cybersecurity for Networked Medical Devices Containing Off-the-Shelf (OTS) Software.
MDCG 2019-16—Guidance on Cybersecurity for Medical Devices.
Australia TGA, Guidance on Personalized Medical Devices (including 3D-printed Devices) regulatory reforms. 2022.
Health Canada, Supporting Evidence for Implantable Medical Devices Manufactured by 3D Printing. Apr 2019.
China NMPA, Technical Review Guidance for the Registration of Personalized Additive Manufacturing Medical Devices of Passive Implantable Bone, Joint and Oral Hard Tissues.
Japan MHLW, Guidance on Evaluation of Customized Orthopedic Devices for Osteosynthesis. Dec 2010.
Japan MHLW, Guidance on Evaluation of Orthopedic Customized Artificial Hip Joint Prosthesis. Dec 2011.
Singapore HSA, Regulatory Guideline for 3D-Printed Medical Devices, July 2021
South Korea MFDS, Guidance for Patient-matched Medical Devices manufactured using 3D printers. Dec 2015.
Discussion Papers. AI in Drug Manufacturing, (March 2023);
Using Artificial Intelligence and ML in the Development of Drug and Biological Products. FDA, CDER. May 2023.
Reflection paper on the use of Artificial Intelligence (AI) in the medicinal product lifecycle. EMA, CVMP & CHMP. Jul 2023.
GMLP for Medical Device Development: Guiding Principles. MHRA, Health Canada, FDA. Oct 2021.
Documentos guías publicados por IMDRF / GHTF
GHTF/SG3/N99-10:2004 (Edition 2) Quality Management Systems – Process Validation Guidance.
GHTF/SG1/N71:2012 Definitions of the Terms ‘Medical Device’ and ‘In Vitro Diagnostic (IVD) Medical Device’.
GHTF/SC/N4:2012 (Edition 2) Glossary and definition of terms used in GHTF documents.
IMDRF Principles and Practices for Medical Device Cybersecurity.
Documentos guías publicados por la industria
ISPE. GAMP 5 Guide 2nd Edition. A Risk-Based Approach to Compliant GxP Computerized Systems. July 2022.
ISPE. GAMP Good Practice Guide: Enabling Innovation. Sep 2021.
ISPE. RDI GAMP Guides: Records and data integrity.
AAMI TIR45:2023; Guidance on the use of agile practices in the development of medical device software.
¿Qué ofrece Trescal?
Desde TRESCAL ofrecemos servicios de soporte relativos al software de dispositivo médico según las normativas de aplicación, ofreciendo consultoría de calidad específica en las siguientes materias:
Auditoría de software.
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Proceso de desarrollo y mantenimiento del software de dispositivo médico (IEC 62304).
Desarrollo del VMP / VP.
Establecimiento de estrategias.
Desarrollo de los expedientes de validación.
Actividades de verificación y validación.
Transición a enfoque CSA (Computer Software Assurance).
Cualificacion de Infraestructura IT
Evaluación de proveedores de SW / tecnológicos.
Acompañamiento a proveedores en pruebas de aceptación.
Procedimientos del sistema de gestión de calidad.
Actividades de formación dirigida.
TRESCAL es un proveedor global de servicios para la industria LIfe Sciences que ofrece servicios de implantación/consultoría en sistemas de calidad de productos sanitarios, gestión de riesgos, Data Integrity, validación de sistemas, cualificación de equipos, calibraciones (incluyendo calibraciones dentro del sector médico/sanitario). Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del proyecto de validación, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Acrónimos
AI: Artificial Intelligence
CDRH: Center for Devices and Radiological Health
CSA: Computer Software Assurance
FDA: Food and Drug Administration
GHTF: Global Harmonization Task Force
IMDRF: international Medical Device Regulators Forum
ML: Machine Learning
PMS: Programmable Electrical Medical Systems
RTM: Requirements Traceability Matrix
SME: Subject-Matter Expert
SW: Software
Imagen1
SW de dispositivos médicos / producto sanitario: ciclo de vida y validación. Parte 1
Introducción
La rápida evolución en la tecnología existente está revolucionando la forma en la que nos comunicamos y gestionamos la información. La digitalización de los dispositivos médicos para dotarlos de tecnologías avanzadas clave de la industria 4.0. no es un asunto desconocido dentro del sector de dispositivos médicos, en donde el uso de dispositivos médicos inteligentes es ya una clara apuesta y tendencia del sector. Se estima que uno de cada cuatro dispositivos médicos incorpora software para dispositivos médicos o son dispositivos médicos por sí mismos.
Por otro lado, la digitalización de la atención sanitaria está permitiendo a pacientes y médicos interactuar con la información de salud de una manera sin precedentes para su mejor optimización y sostenibilidad. Como ejemplos clave se encuentran la tecnología Cloud, Big Data, IoT o AI/ML.
A su vez, este proceso de digitalización está generando una enorme cantidad de datos que supera con creces las capacidades analíticas de los médicos a nivel individual, por lo que estamos viviendo un gran auge de herramientas de inteligencia artificial como catalizador del análisis de datos. Además, no debemos olvidarnos de la gran eclosión del uso generalizado de teléfonos inteligentes y productos digitales portátiles.
El software se ha convertido en consecuencia en una parte importante del panorama del campo de los dispositivos médicos o productos sanitarios, y debemos implementarlo de una forma segura y efectiva para asegurar el uso y seguridad del dispositivo médico en cuestión.
Definiciones de SW relacionado con dispositivos médicos o productos sanitarios
A nivel mundial, existen muchos mercados para la comercialización de dispositivos médicos, pero la Unión Europea y Estados Unidos son quizás los de mayor relevancia al ser los más grandes.
Es importante introducir y remarcar que las regulaciones de la Unión Europea y de los Estados Unidos no utilizan los mismos términos en referencia al SW relacionado con dispositivos médicos. Por un lado, la Unión Europea habla de MDSW (Medical Device Software), y Estados Unidos de SaMD (Software as a Medical Device).
Aclarar que el término SaMD es un concepto internacional, según lo define el International Medical Device Regulators Forum (IMDRF), y que éste se incluye en la legislación de muchos países (a parte de la de Estados Unidos, como es por ejemplo la del Reino Unido, o la del Canadá), mientras que MDSW es exclusivamente un término utilizado por la Unión Europea.
A continuación, revisamos y resumimos cada enfoque, y los matices más relevantes a tener en cuenta:
Estados Unidos: SaMD, SiMD
Para la FDA el software relacionado con los dispositivos médicos puede ser catalogado dentro de las siguientes tipologías:
Software como Dispositivo Médico (SaMD): Software que por sí solo es un dispositivo médico. Es decir, es aquel que «está destinado a ser utilizado para uno o más fines médicos sin ser parte de un dispositivo de hardware«. La FDA define SaMD utilizando la descripción presentada por el International Medical Device Regulators Forum (IMDRF), de la que es miembro. Para ello, debemos conocer bien el uso previsto del producto y sus indicaciones de uso, y estar en conformidad con la definición de dispositivo de la 181 sección 201(h) de la FD&C Act.
Software en un Dispositivo Médico (SiMD): Software que es parte integral de un dispositivo médico que es utilizado para operar o controlar un dispositivo de hardware pero que no cumple con los requisitos para ser categorizado como SaMD. Es decir, es aquel que ayuda a ejecutar un dispositivo médico de hardware (por ejemplo, alimentando su mecánica o produciendo una interfaz gráfica). Sin este software, el dispositivo no podría funcionar correctamente. Un “software embebido” o “firmware” que forma parte de un dispositivo médico de hardware, o un accesorio SW para el control remoto o pantalla del dispositivo médico hardware no cumpliría con la definición de SaMD.
Software utilizado en la fabricación o mantenimiento de un dispositivo médico.
A continuación, revisamos unos ejemplos de cuándo un software es SaMD y no lo es:
Software como Dispositivo Médico (SaMD)
Software que proporciona información (input) a otro dispositivo médico con hardware o software médico diferente. Por ejemplo, el software de planificación de tratamientos que suministra información utilizada en un acelerador lineal.
El software con uso previsto médico que opera en una plataforma hardware convencional (ej.: ordenador, tableta, móvil). Por ejemplo, el software destinado al diagnóstico de una enfermedad.
El software que está conectado a un dispositivo médico que contiene hardware pero que no es necesario para que ese producto alcance su uso previsto.
Software que no es un Dispositivo Médico (SaMD)
Software utilizado para «accionar o controlar» los motores y la bomba de infusión de medicamentos, o software que controla un marcapasos (SiMD).
Software requerido por un dispositivo médico de hardware para realizar el uso previsto del dispositivo médico de hardware, incluso si se vende por separado del dispositivo médico de hardware (SiMD).
Software que se basa en datos de un dispositivo médico, pero que no tiene un propósito médico, por ejemplo, software que cifra datos para su transmisión desde un dispositivo médico.
Software que permite la comunicación clínica y el flujo de trabajo, incluido el registro de pacientes, la programación de visitas, las llamadas de voz y las videollamadas.
Software que monitoriza el rendimiento o el funcionamiento adecuado de un dispositivo con el fin de darle servicio (por ejemplo, software que monitoriza el rendimiento del tubo de rayos X para anticipar la necesidad de reemplazo), o software que integra y analiza datos de control de calidad de laboratorio para identificar errores aleatorios o tendencias en la calibración de los IVDs.
Software que proporciona parámetros que se convierten en entradas (inputs) para el software como dispositivo médico pero que no tiene un propósito médico. Por ejemplo, una base de datos que incluye funciones de búsqueda y consulta por sí misma.
En el ámbito de la Unión Europea el software de dispositivo médico (MDSW) es el software destinado a ser utilizado, solo o en combinación, para un propósito especificado en la definición de «dispositivo médico» conforme a la MDR o IVDR, independientemente que el software sea independiente o impulse o influya en el uso de un dispositivo.
En resumen, se deben cumplir las siguientes tres condiciones para que el software sea clasificado como MDSW:
Tener un propósito médico por sí solo,
Realizar una acción sobre datos más allá del almacenamiento, archivo, comunicación, búsqueda simple o compresión sin pérdidas, y
Estar destinado al beneficio de pacientes individuales.
Cabe mencionar que los siguientes tipos de software que no tienen un propósito de dispositivo médico en sí mismos y, por lo tanto, no se consideran MDSW, aún están sujetos a los requisitos del EU MDR o IVDR, según corresponda:
Software que corresponde a la definición legal de «accesorio» de un dispositivo médico o de un dispositivo de diagnóstico in vitro (IVD),
Software que corresponde a productos sin fines médicos, según lo enumerado en el Anexo XVI del MDR de la EU,
Software que impulsa o influye en el uso de un dispositivo médico (hardware) o IVD, siempre que no tenga un propósito médico separado y no cree información por sí solo (en cuyo caso, el módulo de software con un propósito médico separado o que genere información cualificaría como MDSW). Este software puede, entre otros ejemplos:
(a) operar, modificar el estado o controlar el dispositivo ya sea a través de una interfaz (por ejemplo, software, hardware) o a través del operador de este dispositivo
(b) o suministrar salida relacionada con el funcionamiento (hardware) de ese dispositivo
MDCG 2023-4: Medical Device Software (MDSW) – Hardware combinations Guidance on MDSW intended to work in combination with hardware or hardware components
Manual: Manual on borderline and classification under Regulations (EU) 2017/745 and 2017/746
Infografía: Is your software a Medical Device?
MDCG 2021-24: Guidance on classification of medical devices
MDCG 2020-1: Guidance on clinical evaluation (MDR) / Performance evaluation (IVDR) of medical device software.
MDCG 2019-16: Guidance on cybersecurity for medical devices
MDCG 2019-11: Qualification and classification of software – Regulation (EU) 2017/745 and Regulation (EU) 2017/746.
Diferencias entre MDSW y SaMD
Mientras que SaMD se refiere únicamente al software que es independiente y excluye completamente a cualquier software “embebido” en un dispositivo médico físico, MDSW puede ser independiente o parte de un dispositivo médico de hardware, en el caso de que tenga su propio propósito de dispositivo médico además del propósito de impulsar/influir en el dispositivo médico de hardware. En MDCG 2019-11 se proporcionan un par de ejemplos de software “embebido” con fines médicos propios:
Software de análisis de imágenes de melanoma destinado a controlar un escáner de luz láser de infrarrojo cercano.
MDSW pretende medir y transmitir los niveles de glucosa en sangre, calcular la dosis de insulina necesaria.
Por otro lado, el término de software “standalone” (o independiente) utilizado ampliamente en la industria de la tecnología médica no es sinónimo de MDSW «standalone» ya que un MDSW «standalone» aún puede controlar o influir en un dispositivo médico de hardware.
Ciclo de vida del SW de dispositivos médicos: IEC 62304
La IEC 62304 es una norma internacional que establece requisitos y directrices para el ciclo de vida del software de dispositivos médicos. Esta norma es un estándar de consenso reconocido por las agencias de salud que establece la seguridad y eficacia de un dispositivo médico que contiene software.
El objetivo principal de la IEC 62304 es proporcionar un marco de trabajo para el desarrollo y mantenimiento de software en dispositivos médicos, asegurando la seguridad y eficacia de estos productos. La norma se enfoca en la gestión de riesgos, la documentación, la validación y verificación del software, y garantiza que se cumplan los requisitos reglamentarios y de calidad propios de la industria de dispositivos médicos.
Por otro lado, la IEC 62304 no cubre la parte de validación clínica del sistema SW como dispositivo médico, sino la parte técnica, no clínica, desde la liberación hasta el mantenimiento y discontinuación.
Según este estándar los procesos del ciclo de vida del software de dispositivo médico son los siguientes:
Desarrollo del software.
Mantenimiento del software.
Gestión de riesgos del software.
Gestión de la configuración del software.
Resolución de problemas del software.
Desarrollo del software
El proceso de desarrollo del software está comprendido por las siguientes etapas:
Plan de desarrollo del software
Análisis de requisitos del software.
Diseño arquitectónico del software.
Diseño detallado del software.
Implementación y verificación de la unidad software.
Integración y pruebas del sistema de software.
Liberación de software.
Pero antes de iniciar el proceso de desarrollo del SW, los fabricantes de dispositivos médicos deben realizar una clasificación de la seguridad del software para la asignación de una clase (A, B o C) a cada sistema de software que se implemente en el dispositivo médico según su severidad conforme a los posibles efectos adversos que puedan causar sobre el paciente. Esta severidad puede ser la siguiente:
Clase A: no es posible que se produzcan lesiones ni daños a la salud.
Clase B: es posible que se produzcan lesiones o daños no graves a la salud.
Clase C: es posible la muerte o lesiones graves.
El propósito de realizar esta clasificación de seguridad del software sirve de base para determinar qué tan riguroso debe ser el proceso de desarrollo de software posterior (ver figura 1):
Capítulo
Clase A
Clase B
Clase C
Planificación del desarrollo (5.1.)
x
x
x
Análisis de los requisitos (5.2)
x
x
x
Diseño arquitectura (5.3)
x
x
Diseño detallado (5.4)
x
x
Implementación (5.5)
x
x
x
Ensayos test unitarios (5.5)
x
x
Integración (5.6)
x
x
Ensayos de integración (5.6)
x
x
Ensayos del sistema (5.7)
x
x
x
Liberación (5.8)
x
x
x
Figura 1: Actividades recogidas en 62304 en función del riesgo del dispositivo médico
Por otro lado, tener en cuenta que aunque la IEC 62304 tiene una estructura bastante lineal es posible a su vez cumplir con sus requisitos utilizando un desarrollo de SW ágil. De hecho, existe otro estándar AAMI TIR 45 que ofrece orientación sobre el uso de prácticas ágiles en el desarrollo de software de dispositivos médicos.
Es por ello importante la definición de un modelo de ciclo de desarrollo del software acorde a los estándares actuales que pueda tener cabida a enfoques agiles de desarrollo de SW más flexibles, dinámicos e incrementales, especialmente ante sistemas de software complejos en donde se necesita un mayor ajuste y redefinición de requisitos.
Mantenimiento del software
Debe establecerse un plan de mantenimiento que describe cómo se gestionará el mantenimiento del software a lo largo de su ciclo de vida. Esto incluye la identificación de las actividades de mantenimiento necesarias, la frecuencia de las actualizaciones, la gestión de riesgos asociados con el mantenimiento, y la asignación de recursos y responsabilidades.
El proceso de mantenimiento del software descrito en IEC 62304 consta de los siguientes puntos:
Establecimiento del plan de mantenimiento del software
Análisis de problemas y modificaciones.
Implementación de las modificaciones.
Gestión de riesgos del software
Los requisitos de gestión de riesgos de la IEC 62304 se correlacionan con los de ISO 14971, norma internacional sobre la aplicación de la gestión de riesgos en dispositivos médicos, por lo que resulta fácil notar su interconexión. Este proceso implica la identificación, análisis, evaluación y gestión de los riesgos asociados con el software médico a lo largo de todo su ciclo de vida. Esto incluye, por ejemplo:
Análisis del software en términos de contribución a situaciones peligrosas.
Medidas de control de riesgo.
Verificación de las medidas de control de riesgo.
Gestión de riesgos de cambios en el software.
Gestión de la configuración del software
Este proceso se encarga de mantener un control estricto de las configuraciones y versiones del software a lo largo de su ciclo de vida permitiendo su trazabilidad. De esta forma se identifican y definen los elementos de software (incluyendo la documentación), así como se controlan los cambios y liberaciones, documentando e informando del estado de los elementos de configuración y solicitudes de cambio. Se incluye:
Identificación de la configuración: Identificación de cada elemento de configuración del software y sus versiones.
Control de Cambios: Incluyendo el proceso de solicitud de cambios.
Histórico del control de los elementos de configuración.
Resolución de problemas del software
Finalmente, el proceso de resolución de problemas del software implica la identificación y gestión de problemas, errores o no conformidades en el software, así como la implementación de acciones correctivas para su resolución. Este proceso proporciona un medio oportuno, responsable y documentado para garantizar que el problema es analizado y resuelto.
IEC 62304 establece un esquema general para un proceso de resolución de problemas:
Elaboración de los informes de problemas
Estudio del problema
Aviso a las partes interesadas
Uso del proceso de control de cambios
Mantenimiento de los registros
Análisis para la detección de tendencias en los informes de problemas
Verificación de la resolución de problemas de software
Contenido de la documentación de prueba/ensayo
Como regla general, es probable que encuentre menos problemas con el tiempo a medida que su producto madure. Sin embargo, es probable que la gravedad de los problemas que encuentre más adelante sea aún mayor y requiera un proceso más riguroso para resolución.
Normativas de referencia
Las empresas fabricantes de software de dispositivos médicos deben disponer de un sistema de gestión de calidad adecuado que asegure el correcto ciclo de vida del dispositivo médico. A continuación, se dan como referencia y orientación las siguientes guías y estándares:
Estándares
ISO 13485 Medical Devices – Quality Management Systems – Requirements for Regulatory Purposes
ISO 14971 Medical Devices – Application of Risk Management to Medical Devices
ISO 14155 Clinical Investigation of Medical Devices for Human Subjects: Good Clinical Practice
IEC 62366-1 Medical devices – Part 1: Application of usability engineering to medical devices
IEC 62304:2006 Medical device software. Software life cycle processes.
IEC 82304-1:2016 Health software – Part 1: General requirements for product safety
ISO/TS 82304-2:2021 Health software – Part 2: Health and wellness apps. Quality and reliability
IEC 81001-5-1 IEC 81001-5-1 Health software and health IT systems safety, effectiveness and security.
ISO/IEC TR 24028:2020 Information technology — Artificial intelligence — Overview of trustworthiness in artificial intelligence
ISO/IEC DIS 5259-1 Artificial intelligence — Data quality for analytics and machine learning (ML) Under development (draft 2023)
Documentos guías publicados por Autoridades Regulatorias
CDRH. Draft Guidance – Computer Software Assurance for Production and Quality System Software. Sep 2022.
Europe MDCG 2021-3, Questions and Answers on Custom-Made Devices (& considerations on Adaptable medical devices and Patient-matched medical devices). Mar 2021.
US FDA 21 CFR 820.30, Design Control Guidance for Medical Device Manufacturers. Mar 1997.
US FDA CDRH, Technical Considerations for Additive Manufactured Devices – Guidance for Industry and Food and Drug Administration Staff. Dec 2017.
US FDA CDRH, Applying Human Factors and Usability Engineering to Medical Devices – Guidance for Industry and Food and Drug Administration Staff. Feb 2016.
US FDA CDER, CBER – Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients – Guidance for Industry (Revision 1). Sept 2016.
MITRE_MDIC (FDA) – Playbook for Threat Modeling Medical Devices.
Cybersecurity for Networked Medical Devices Containing Off-the-Shelf (OTS) Software.
MDCG 2019-16—Guidance on Cybersecurity for Medical Devices.
Australia TGA, Guidance on Personalized Medical Devices (including 3D-printed Devices) regulatory reforms. 2022.
Health Canada, Supporting Evidence for Implantable Medical Devices Manufactured by 3D Printing. Apr 2019.
China NMPA, Technical Review Guidance for the Registration of Personalized Additive Manufacturing Medical Devices of Passive Implantable Bone, Joint and Oral Hard Tissues.
Japan MHLW, Guidance on Evaluation of Customized Orthopedic Devices for Osteosynthesis. Dec 2010.
Japan MHLW, Guidance on Evaluation of Orthopedic Customized Artificial Hip Joint Prosthesis. Dec 2011.
Singapore HSA, Regulatory Guideline for 3D-Printed Medical Devices, July 2021
South Korea MFDS, Guidance for Patient-matched Medical Devices manufactured using 3D printers. Dec 2015.
Discussion Papers. AI in Drug Manufacturing, (March 2023);
Using Artificial Intelligence and ML in the Development of Drug and Biological Products. FDA, CDER. May 2023.
Reflection paper on the use of Artificial Intelligence (AI) in the medicinal product lifecycle. EMA, CVMP & CHMP. Jul 2023.
GMLP for Medical Device Development: Guiding Principles. MHRA, Health Canada, FDA. Oct 2021.
Documentos guías publicados por IMDRF / GHTF
GHTF/SG3/N99-10:2004 (Edition 2) Quality Management Systems – Process Validation Guidance.
GHTF/SG1/N71:2012 Definitions of the Terms ‘Medical Device’ and ‘In Vitro Diagnostic (IVD) Medical Device’.
GHTF/SC/N4:2012 (Edition 2) Glossary and definition of terms used in GHTF documents.
IMDRF Principles and Practices for Medical Device Cybersecurity.
Documentos guías publicados por la industria
ISPE. GAMP 5 Guide 2nd Edition. A Risk-Based Approach to Compliant GxP Computerized Systems. July 2022.
ISPE. GAMP Good Practice Guide: Enabling Innovation. Sep 2021.
ISPE. RDI GAMP Guides: Records and data integrity.
AAMI TIR45:2023; Guidance on the use of agile practices in the development of medical device software.
¿Qué ofrece Trescal?
Desde TRESCAL ofrecemos servicios de soporte relativos al software de dispositivo médico según las normativas de aplicación, ofreciendo consultoría de calidad específica en las siguientes materias:
Auditoría de software.
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Proceso de desarrollo y mantenimiento del software de dispositivo médico (IEC 62304).
Desarrollo del VMP / VP.
Establecimiento de estrategias.
Desarrollo de los expedientes de validación.
Actividades de verificación y validación.
Transición a enfoque CSA (Computer Software Assurance).
Cualificacion de Infraestructura IT
Evaluación de proveedores de SW / tecnológicos.
Acompañamiento a proveedores en pruebas de aceptación.
Procedimientos del sistema de gestión de calidad.
Actividades de formación dirigida.
TRESCAL es un proveedor global de servicios para la industria LIfe Sciences que ofrece servicios de implantación/consultoría en sistemas de calidad de productos sanitarios, gestión de riesgos, Data Integrity, validación de sistemas, cualificación de equipos, calibraciones (incluyendo calibraciones dentro del sector médico/sanitario). Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del proyecto de validación, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Acrónimos
AI: Artificial Intelligence
EC: European Comission
EU: European Union
FDA: Food and Drug Administration
GHTF: Global Harmonization Task Force
IMDRF: international Medical Device Regulators Forum
IoT: Internet of Things
IVD: in vitro diagnostic medical device
IVDR: In Vitro Medical Device Regulation (EU) 2017/746
MDCG: Medical Device Coordination Group
MDR: Medical Device Regulation (EU) 2017/745
MDSW: Medical Device Software
ML: Machine Learning
SaMD: Software as a Medical Device
SiMD: Software in Medical Device
SW: Software
GMP Qualification and validation
PUNTOS CLAVE PARA AFRONTAR EL NUEVO ANEXO 1 EU GMP
El nuevo anexo 1 representa un reto y un buen recordatorio para los fabricantes de productos estériles (y no estériles) en la mejora del sistema PQS (Pharmaceutical Quality System), para su mejor alineación en materia de control y prevención de la contaminación del producto final. Como cualquier otro proyecto de mejora, debemos planificar y estructurar las diferentes actividades a llevar a cabo para la consecución de los nuevos objetivos de forma óptima y efectiva, en búsqueda de una mejor conformidad de las instalaciones y procesos de fabricación.
Para llevar a cabo esta andadura con éxito es primordial tener en cuenta los siguientes aspectos:
Creación de un equipo multifuncional experto en la materia. Debe existir un adecuado trabajo de equipo y una buena comunicación desde el inicio del proyecto entre todas las partes y departamentos involucrados (ej.: departamento de microbiología, producción, garantía de calidad, control de calidad, validaciones, regulación, mantenimiento), incluyendo expertos en ciencias de los datos, gestión de los riesgos, y proveedores de servicios críticos. En este punto, debemos revisar bien nuestros procesos para delimitar alcances iniciales y responsabilidades.
Conocer el nivel de madurez en materia de control de contaminación, es decir conocer si disponemos de experiencia amplia en fabricación de medicamentos estériles, u otros productos en donde el control de la contaminación es un punto importante.
Recopilación de datos relevantes. Debe realizarse un esfuerzo colectivo de gestión del conocimiento para mejorar y actualizar el conocimiento que se encuentra en documentos y bases de datos. Para ello, recopilaremos toda la información técnica del producto y del proceso, así como nos rodearemos de toda la documentación interna en materia de control de contaminación.
Realizar un análisis de brechas de los cambios requeridos del Anexo 1. En el caso de instalaciones ya existentes, el realizar un Gap Assessment para la identificación de brechas en las novedades del anexo 1, es un ejercicio altamente recomendable para saber a priori qué nivel de brecha tenemos entre la situación actual y la definida por el anexo 1.
Prestar atención al diseño y control de las instalaciones, equipos, servicios y procesos. La evaluación del diseño de la instalación y de los procesos (e incorporación de nuevas tecnologías de fabricación avanzadas, sistemas de barrera -RABS, aisladores-, y nuevas metodologías de monitorización/control), así como la revisión del estado de su estado de cualificación / validación, es fundamental para garantizar que las prácticas actuales cumplen con los nuevos requisitos.
Prestar atención al personal. Los procesos y prácticas en materia de prevención y control de la contaminación pueden requerir cambios dependiendo de la interpretación e implementación previa de los requisitos del Anexo 1. Debe existir formación específica en materia de contaminación (ej.: prácticas de vestimenta, higiene, trabajo aséptico, desinfección y limpieza), así como requerimientos de cualificación y descualificación del personal.
Trabajar con proveedores expertos. Rodearnos de proveedores expertos nos permite una mayor garantía de todos los puntos del proceso de fabricación y cadena de suministro. Su evaluación, así como la elaboración de acuerdos de calidad específicos, son asuntos clave a tener en consideración.
PQS efectivo y cierre de la política CCS Contamination Control Strategy). Debe existir una alineación del CCS con el sistema PQS para la evaluación holística de los riesgos, y medidas de contención existentes. Evaluar el impacto de los cambios del Anexo 1 en los aspectos definidos del CCS debe conllevar una revisión periódica continua del PQS. Es importante el análisis de los datos y tendencias en continuo para el establecimiento de los planes de mejora y mitigación oportunos. La disposición de herramientas de análisis e investigación avanzadas y sólidas juegan aquí un papel relevante.
Sistema QRM (Quality Risk Management) robusto. El contar con un sistema de gestión de los riesgos para la calidad robusto es crucial para el efectivo proceso de toma de decisión basado en los riesgos. Es importante disponer de los datos adecuados, así como elegir la herramienta de gestión de riesgos y el nivel de formalidad del proceso QRM más adecuado en cada etapa del proyecto en base al nivel de nivel de incertidumbre, complejidad, e importancia.
Estrategia de control de contaminación y PQS
Conclusión
Los fabricantes deben adoptar un enfoque basado en el riesgo para analizar sus procesos e instalaciones actuales para la determinación de las medidas necesarias más efectivas para cumplir con los requisitos del nuevo anexo 1 y así mejorar su PQS en términos de control y prevención de la contaminación. Y todo ello, sobre una base de revisión periódica y continua.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, validación de sistemas, cualificación de equipos y calibración. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten:
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Gap análisis de la estrategia actual de control de la contaminación.
Evaluación de proveedores.
Cualificación de salas limpias.
Ensayos de direccionalidad de humo (visualización y filmación)
Cualificación de servicios farmacéuticos.
Cualificación de equipos de esterilización.
Establecimiento y mantenimiento de los planes de verificación de salas limpias.
Desarrollo / revisión de la estrategia de control de la contaminación (CCS).
Validaciones de proceso.
Validaciones de limpieza.
Desarrollo de los expedientes de validación / cualificación.
Acompañamiento a proveedores en pruebas de aceptación.
Revisiones periódicas.
Calibración instrumental.
Actividades de formación dirigida.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar la correcta alineación de su sistema de calidad, procesos e instalaciones con los nuevos requisitos del Anexo 1, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
IMG-20221005-WA0014
TRESCAL PARTICIPARÁ EN FARMAFORUM LOS DÍAS 20 y 21 DE SEPTIEMBRE DE 2023
Madrid, 1 de septiembre, 2023. El grupo Trescal estará presente en Farmaforum la feria de referencia del sector farmacéutico el próximo mes de septiembre.
Con el objetivo de seguir ampliando mercado e impulsar la imagen Trescal en el sector life sciences Trescal participará con el stand F27 en Farmaforum, Madrid. La feria tendrá lugar los días 20 y 21 de septiembre en IFEMA, Pabellón 14.
Un año más una oportunidad única para mantenerse al día sobre las últimas actualidades GxP y de los servicios más actuales.
A continuación, detallamos los talleres que ofrecerán:
20 de septiembre de 12 a 14h: Digitalización, novedades de la guía Gamp5 2nd Edition, y relevancia del proveedor tecnológico
Ponente: Mar Díaz, Responsable Técnica Validaciones
Ubicación: sala talleres Farmaforum
Puedes apuntarte a los mismos mediante el siguiente enlace
20 de septiembre a las 12:45: Mesa Redonda «Del Diseño al éxito»
Ponente: Roberto Español, Responsable Técnico Life Sciences
Formação mínima de 12ºano ou curso profissional com experiência comprovada;
Experiência de 3 anos na atividade de ensaios em Ambientes Limpos;
Valorizamos os conhecimentos de Inglês e/ou Castelhano;
Conhecimentos de MS Office, especialmente em EXCEL;
Carta de condução tipo B;
Flexibilidade horaria e disponibilidade para deslocações nacionais.
REQUISITOS DESEJADOS
Organização e gestão do tempo;
Planificação;
Iniciativa e proatividade;
Facilidade de comunicação.
CAPACIDADES NECESSÁRIAS
Rigor e Sentido crítico;
Trabalho em equipa;
Ágil adaptabilidade.
O QUE OFERECEMOS
Salário adequado à experiência e competência demonstrada;
Seguro de saúde;
Oportunidade de crescimento numa grande multinacional.
AI. Artificial intelligence concept. Abstract wireframe digital human face on streaming matrix digital binary code background. Human head in robot digital computer interpretation. Vector illustration.
Nuevo documento de reflexión de la EMA sobre el uso de AI el en el ciclo de vida del medicamento
La Agencia Europea de Medicamentos (EMA, por sus siglas en inglés) ha publicado recientemente un documento borrador sobre el uso de la inteligencia artificial (AI) en el desarrollo y regulación de los medicamentos de uso humano y veterinarios.
Introducción
Este pasado 19 de julio, la EMA ha publicado un documento borrador (Reflection paper) sobre el uso de la AI en el ciclo de vida del medicamento. El documento está actualmente y hasta el 31 de diciembre, en trámite de consulta pública.
Este documento de reflexión quiere brindar consideraciones sobre el uso de AI (Artificial Intelligence) y ML (Machine Learning) en el ciclo de vida de los medicamentos, desde su descubrimiento, hasta el momento posterior a su autorización. Dado el rápido desarrollo de este campo, el objetivo de este documento es reflexionar sobre los principios científicos que son relevantes para la evaluación regulatoria, cuando estas tecnologías emergentes se aplican para apoyar el desarrollo y uso seguro y eficaz de los medicamentos.
Reconocimiento de nuevas oportunidades y desafíos
El nuevo borrador reconoce que este nuevo campo/tecnología emergente representa una gran oportunidad para la industria para mejorar todas las fases del ciclo de vida del medicamento, pero admite que trae a su vez desafíos, como la comprensión de los algoritmos o los posibles fallos técnicos que pudieran surgir, o el uso de datos de entrenamiento no representativos y equilibrados teniendo en cuenta todas las bases relevantes para la no discriminación de poblaciones (introducción de sesgos), la no protección de datos personales, o la falta de integridad de los datos. Por ello, el borrador de la EMA destaca que el desarrollo de la AI debe estar permanentemente guiado por humanos y cumplir con los requisitos legales y éticos pertinentes.
A su vez, en varios aspectos, como son la gestión y gobernanza de datos, el rigor estadístico, los principios normativos, y las mejores prácticas establecidas son directamente aplicables a la AI/ML, por lo que deben realizarse esfuerzos en todas las organizaciones para integrar la competencia de la ciencia de los datos dentro del desarrollo de medicamentos y la farmacovigilancia.
El objetivo es poder garantizar la seguridad de los pacientes y de los resultados con el fin de promover la confiabilidad de la AI/ML.
En resumen, el uso de AI en el ciclo de vida del medicamento debe estar en cumplimiento con los requisitos legales existentes, considerando la ética y sus principios subyacentes, con el debido respeto a los derechos fundamentales.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de infraestructura IT. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del ciclo de vida de su sistema informático e infraestructura IT, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
TRESCAL PARTICIPARÁ EN FARMAFORUM LOS DÍAS 25 y 26 DE SEPTIEMBRE DE 2024
Madrid, 2 de septiembre, 2024. El grupo Trescal estará presente en Farmaforum la feria de referencia del sector farmacéutico este mes de septiembre.
Con el objetivo de seguir ampliando mercado e impulsar la imagen Trescal en el sector life sciences Trescal participará con el stand E32 en Farmaforum, Madrid. La feria tendrá lugar los días 25 y 26 de septiembre en IFEMA, Pabellón 9.
Un año más una oportunidad única para mantenerse al día sobre las últimas actualidades GxP y de los servicios más actuales.
A continuación, detallamos los talleres que ofrecerán:
25 de septiembre de 10 a 12h: Novedades del Anexo I
Ponente: Mar Díaz, Responsable Técnica Validaciones y Roberto Español, Responsable Técnico Trescal Life Sciences
¡Estaremos esperándote en nuestro stand! Puede conseguir invitaciones en el siguiente enlace
TECHNICAL_BNL_ANVERS_2021_PRESSURE (3)
La UPC y el grupo TRESCAL firman un acuerdo para fomentar las vocaciones STEAM entre los jóvenes
La UPC y el grupo TRESCAL, han firmado un convenio de colaboración a través del cual se apoyará a la organización, la gestión y la difusión del programa de Fomento de Vocaciones y Divulgación STEAM de la Universidad, dirigido en los centros educativos de Cataluña.
El convenio de colaboración entre grupo TRESCAL, y la Universitat Politècnica de Catalunya – BarcelonaTech (UPC) se materializará en una aportación de 5.000 euros por parte de la empresa destinados al programa de fomento de vocaciones STEAM de la UPC, a través del cual la Universidad ofrece un catálogo de actividades de fomento de vocaciones STEAM, dirigido principalmente a alumnado de primaria, ESO y bachillerato de centros educativos de Cataluña, así como a familias y a la sociedad en general.
Las actividades que organiza la UPC están orientadas a despertar vocaciones en los estudios STEAM (por sus siglas en inglés de ciencias, tecnología, ingeniería, artes –que incluye arquitectura, urbanismo y edificación– y matemáticas). El programa de actividades está orientado a romper los estereotipos de género y los asociados a las profesiones STEAM, explicar la aplicabilidad social de estas profesiones y dar visibilidad a la actividad de los investigadores y las investigadoras de la Universidad que llevan a cabo estas actividades.
Por su parte, el grupo TRESCAL tiene interés en patrocinar acciones en el ámbito de la educación universitaria con el fin de promover y apoyar acciones y actividades de fomento de vocaciones en ámbitos de especialización de la compañía en el marco de su programa de responsabilidad social corporativa.
La UPC tiene una amplia trayectoria en el desarrollo de proyectos y actividades orientados al fomento de las vocaciones STEAM a través de un amplio programa de actividades dirigido a los centros educativos, al alumnado de primaria, ESO y bachillerato, a sus familias y a la sociedad en general. La UPC ofrece a los centros educativos un amplio catálogo de conferencias de divulgación científica y tecnológica y talleres STEAM, que llevan a cabo personal docente e investigador de la Universidad. El último curso académico se han impartido 450 conferencias, a las que han asistido más de 15.700 jóvenes, y se han organizado 160 talleres STEAM, que han contado con la participación de más de 3.150 alumnos.
LinkedIn (1)
Trescal participará en ADM Sevilla, 14-16 Mayo
Tenemos el placer de anunciarles que el grupo Trescal estará presente en ADM en Sevilla los próximos días 14 – 16 de mayo 2024, la feria de referencia del sector aeroespacial y defensa en España. Con el objetivo de seguir ampliando mercado e impulsar la imagen Trescal en el sector aeroespacial y defensa Trescal participará con el stand D11 en ADM Sevilla en Sevilla City Office (antiguo FIBES Palacio de Congresos).
Nuevo marco de calidad de datos de la EMA / HMA para la regulación de medicamentos.
En diciembre de 2023, la EMA y la HMA publicaron conjuntamente la primera versión completa del documento sobre Marco de Calidad de Datos para la regulación de medicamentos en la EU. Este documento tiene como objetivo caracterizar, evaluar y garantizar la calidad de datos para el respaldo de la toma de decisiones regulatorias.
Introducción y foco
El progreso en la digitalización y la tecnología de la información ha creado nuevas oportunidades que han contribuido a un panorama cada vez más complejo para la toma de decisiones regulatorias. Por ello, se está reconociendo una creciente necesidad de estandarización y la implementación de un marco que describa la calidad de datos. Este marco permitiría al regulador realizar evaluaciones regulatorias confiables sobre si los datos son apropiados para respaldar la toma de decisiones. Pero, aunque se dispone de nuevos tipos o estándares de datos, todavía son escasas las directrices o métodos para demostrar si dichos datos son adecuados para la toma de decisiones.
Por todo ello, se ha visto necesario por parte de la European Medicines Agency (EMA) y la Heads of Medicines Agencies (HMA) la elaboración de un documento Marco de Calidad de Datos (DQF, por sus siglas en inglés) que brinde orientación y guía para el logro de procedimientos de evaluación de calidad coherentes y consistentes. Este documento de 42 páginas, ha sido publicado en el mes de diciembre de 2023, tras ser actualizado tras los comentarios recibidos durante su versión preliminar de octubre de 2022.
Con el establecimiento de un marco de calidad de datos la EMA y la HMA pretenden:
Mejorar la coherencia en la evaluación de la calidad de datos utilizados por los reguladores.
Facilitar el desarrollo de un enfoque estandarizado para la calidad de datos en todas las fuentes de datos.
Favorecer un uso más sistemático de los datos en la toma de decisiones regulatorias.
Apoyar la confianza de los interesados en los datos que respaldan las decisiones regulatorias.
Este marco va alineado además con el objetivo principal del plan de trabajo del Grupo Directivo Conjunto de la HMA-EMA sobre Big Data que es el de desarrollar pautas para el logro de un proceso fortalecido para la cualificación de datos a través de asesoramiento científico, y promover en los Estados miembros la adopción de registros electrónicos de salud, registros, datos genómicos y la disponibilidad segura de los datos.
Alcance y contenido
El documento es la primera publicación del Marco de Calidad de Datos de la EU (DQF) para la regulación de medicamentos y define principios y procedimientos de alto nivel que se aplican en todo el mandato regulatorio de la EMA. Por ello, brinda un análisis sobre las acciones y métricas de calidad de datos que deben considerarse en diferentes casos de uso e introduce un modelo de madurez para guiar la evolución de la automatización para respaldar la toma de decisiones regulatorias basada en datos.
El objetivo es proporcionar un conjunto de definiciones, principios y directrices para su aplicación de forma coherente en cualquier fuente de datos con el fin de caracterizar, evaluar y asegurar la calidad del conocimiento para la toma de decisiones regulatorias. Este marco está destinado además a abarcar el uso primario y secundario de los datos, así como metadatos e información de respaldo.
Sobre las verificaciones de calidad de datos, estas ocurren en varios pasos a lo largo del proceso de generación de evidencia y pueden incluir ciclos de retroalimentación iterativos tal y como se indica la línea discontinua de la figura 1.
Figura 1: Flujo de trabajo típico de procesamiento de datos en el proceso de generación de evidencia.
Por otra parte, dentro del alcance, el documento quiere hacer reseña del concepto de dato, que define como “cualquier activo de información que representa mediciones u observaciones y que pueden usarse para apoyar la toma de decisiones, directa o indirectamente a través del análisis”, así como del de Calidad de datos que define como “la idoneidad para el propósito de las necesidades de los usuarios en relación con la investigación en salud, la formulación de políticas y la regulación, y que los datos reflejan la realidad que intentan representar.”
Se remarca por lo tanto que la calidad de datos debe ser evaluada desde el punto de vista de su idoneidad para satisfacer las necesidades de los usuarios.
Por otra parte, el documento está dirigido principalmente a la red reguladora de medicamentos de la EU, pero la relevancia del contenido puede ser de interés para una gama más amplia de partes interesadas, como titulares de autorizaciones de comercialización, titulares de fuentes de datos, investigadores y asociaciones de pacientes.
Consideraciones en la implementación del DQF
Por otra parte, el nuevo documento marco proporciona un conjunto de observaciones y recomendaciones guía en la implementación del DQF para el logro de mayores niveles de madurez. El documento destaca las siguientes:
Asegurar la calidad en origen: Al diseñar procesos de recopilación y generación de datos, los aspectos que afectan a la calidad de datos deben abordarse de forma inicial ya que cuanto más se alejan los datos del contexto original, más difícil resulta corregir los problemas. Este asunto es particularmente relevante en el caso de los metadatos, ya que el conocimiento del contexto de generación de datos está presente solo en el momento de la generación.
Disponibilidad de datos maestros (MDM) y datos de referencia: La disponibilidad de MDM y datos de referencia tiene un impacto directo en la calidad de datos. A menudo es un requisito previo para la coherencia de los datos e incluso puede afectar la confiabilidad en algunos escenarios de producción de datos, ya que la información desconectada puede generar información errónea.
Alineación del SGC: Un Sistema de Gestión de Calidad es un enfoque formalizado adoptado por una organización que documenta procesos, procedimientos y responsabilidades para lograr políticas y objetivos de calidad (por ejemplo, Good Clinical Practices [GCP], Good Laboratory Practices [GLP] or Good Manufacturing Practice [GMP]). A su vez, estándares como la familia ISO 9000 definen el SGC en todas las industrias, pero se han desarrollado SGC más específicos para industrias o productos específicos. Dentro del sector Life Sciences podemos resaltar los siguientes estándares:
ISO 14155 y Directiva de la UE 2001/20/EC para GCP (datos de ensayos clínicos)
Quality System Regulation (QSR) e ISO 13485 para dispositivos médicos.
Directiva de la UE 2004/9/EC y 2004/10/EC para GLP (datos de investigación de laboratorio).
ISO 15189 e ISO 17025 (datos de laboratorios clínicos).
Controles sobre los sistemas informáticos: Ante el actual marco de digitalización, los sistemas informáticos utilizados para crear, modificar, mantener, archivar, recuperar o transmitir datos son un elemento crucial a tener en cuenta cuando hablamos de calidad de datos. Debemos por ello, asegurar que disponemos de sistemas informáticos efectivos, confiables y de alta calidad. La validación del sistema informático garantiza que el software se implementa adecuadamente y que existan los controles de proceso necesarios para usarlo de acuerdo a especificaciones, incluyendo la documentación asociada, el control de acceso, la gestión de proveedores y las auditorías. Por otro lado, debemos contar con un ciclo de vida de desarrollo de software que incluya un sistema de garantía de calidad del software para garantizar su diseño, desarrollo y verificación cuando el dato del SW es el propio producto (ej.: software de dispositivo médico).
El papel de los estándares ISO de la industria: La Organización Internacional de Normalización (ISO) ha elaborado normas que proporcionan marcos para la implementación de diversos aspectos de la gestión de datos, que se prueban en el campo y para las cuales se establecen plataformas, servicios de apoyo y organismos de certificación. Estos estándares a menudo se desarrollan para la implementación de industrias donde no se aplica la toma de decisiones regulatorias de la EMA.
ISO 9000: Describe los estándares para los sistemas de gestión de calidad en todos los niveles de una organización. Se podría considerar la adopción de esta norma si no se aplica ningún SGC específico de la industria.
ISO 8000: Describe los estándares de gestión de calidad de datos de una organización sometidos a un ciclo de mejora continua. La ISO 8000-150 especifica los principios fundamentales de la gestión de la calidad de datos maestros, centrada en los procesos, y los requisitos para la implementación, el intercambio de datos y la procedencia.
ISO 25012: Define un modelo general de calidad de datos para datos retenidos en un formato estructurado dentro de un sistema informático, típico de los datos considerados para la toma de decisiones regulatorias. Proporciona un marco para establecer requisitos de calidad de datos, medidas de calidad de datos y un plan para realizar evaluaciones de calidad de datos. En un modelo de Calidad de Datos se establecen las 15 características de Calidad de Datos que se deben tener en cuenta a la hora de evaluar las propiedades de un producto de datos determinado (exactitud, completitud, consistencia, credibilidad, actualidad, accesibilidad, conformidad, confidencialidad, eficiencia, precisión, trazabilidad, comprensibilidad, disponibilidad, portabilidad y recuperabilidad).
ISO 13485: Es la norma que especifica los requisitos de un sistema de gestión de la calidad para que una organización diseñe, construya y obtenga autorización para dispositivos médicos que cumplan sistemáticamente con los requisitos reglamentarios y del cliente.
Consideraciones sobre integridad de datos: ALCOA ++ es un marco para la integridad de los datos utilizado en toda la industria farmacéutica. Postula un conjunto de atributos que deben cumplir los datos y su documentación para que éstos sean Atribuibles, Legibles, Contemporáneos, Originales, y Acertados (ALCOA). El ++ se refiere a los siguientes atributos: Completo, Consistente, Duradero, Disponible, Trazable.Siguiendo esta premisa, debemos centrarnos en los determinantes fundamentales que afectan principalmente las dimensiones de Confiabilidad de los datos, pero también en las de Extensión, Coherencia y Oportunidad.
Figura 2: Atributos ALCOA ++
ATRIBUIBLE: Atribuible a la persona o sistema que genera los datos. Vinculado a la fuente de datos LEGIBLE: Fácilmente legible por ser humano (reúne toda la información para que dato sea completo) CONTEMPORÁNEO: Registrados en el momento de su adquisición ORIGINAL: Primer registro del dato, o “True Copie” que preserve el contenido y significado ACERTADO: Verificado como correcto. Sin error. Verificación de cálculos, algoritmos++ completo, consistente, duradero, disponible, trazable.
Controles sobre la Calidad de datos: Existen diferentes implementaciones posibles de controles de calidad de datos dependiendo si los hechos reales que representan los datos son conocidos y accesibles o no. En caso que los hechos reales sean conocidos, los datos se pueden probar mediante validación (versus los registros fuente que contienen estos hechos), aunque la validación puede ser costosa y consumir mucho tiempo, y a menudo requiere el uso de evaluadores si los hechos no están disponibles en forma estructurada y legible. Alternativamente, los datos se pueden probar mediante métricas de plausibilidad intrínsecas, evaluando el conjunto de datos con respecto a: otros datos del mismo conjunto de datos, diferentes rangos de referencia externos o otras tendencias plausibles (ciertos datos pueden ser válidos cuando se observan individualmente, pero la tendencia colectiva de todos los datos de un tipo debe seguir las distribuciones o tendencias esperadas).
Conclusiones
La calidad de datos es un elemento crucial para realizar todo el potencial de la regulación basada en datos y respalda la confianza de los pacientes y profesionales de la salud. El proyecto de Marco de Calidad de Datos para la regulación de medicamentos en la Unión Europea establece los criterios de calidad para los datos para garantizar que éstos sean aptos para el propósito de respaldar las decisiones de beneficio-riesgo.
Normativas de referencia
European Medicines Regulatory Network Data Standardisation Strategy. December 16th, 2021.
Big Data Steering Group, Big Data Workplan 2022-2025.
Good Practice Guide for the use of the Metadata Catalogue of Real-World Data Sources, EMA/787647/2022.
Guideline on computerised systems and electronic data in clinical trials. EMA/INS/GCP/112288/2023.
PIC/S Guidance. Good practices for data management and integrity in regulated GMP/GDP environments. PI 041-1. 1 July 2021.
Good Practice Guide for the use of the Metadata. Catalogue of Real-World Data Sources. EMA/787647/2022. V 1.0 (draft)
Acrónimos
DQF: Data Quality Framework
EMA: European Medicines Agency
HMA: Heads of Medicines Agencies
EU: European Union
MDM: Master Data Management
SGC: Sistema de Gestión de Calidad
ISO: International Organization for Standardization
EHDS: European Health Data Space
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Sciences que ofrece servicios de implantación/consultoría en sistemas de calidad, gestión de riesgos, Data Integrity, Data Governance, validación de sistemas informáticos, cualificación de equipos, calibraciones. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para adecuar su sistema de gestión de calidad al nuevo marco de calidad de datos, revisando estrategias y controles, o si necesita soporte en la realización de verificaciones y validaciones no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Imagen1-1
SW de dispositivos médicos / producto sanitario: ciclo de vida y validación. Parte 2
Tal y como vimos en el anterior artículo (Parte 1) el software de un dispositivo médico / producto sanitario debe ser implementado de una forma segura y efectiva para asegurar el uso y seguridad del producto final.
El estándar IEC 62304, de aplicación tanto para el software incorporado en un dispositivo, como para el software que es un dispositivo médico en sí mismo, sirve como referencia para todo el ciclo de vida del software de dispositivos médicos.
A continuación, echaremos un vistazo a las actividades de Verificación y Validación(V&V) dentro del proceso de desarrollo del software.
Verificación y validación
Las actividades de verificación y validación (V&V) son fundamentales para la obtención de dispositivos médicos seguros.
Mientras, la verificación se refiere a la comprobación del producto desarrollado conforme a sus requisitos especificados, la validación comprueba si se cumple su uso previsto en arreglo a las necesidades del usuario. De forma general, los términos de verificación y validación responden cada uno, a las siguientes preguntas:
Verificación: ¿El diseño del dispositivo cumple los requisitos definidos?
Validación: ¿Se cumplen los requisitos para el uso previsto del dispositivo? ¿Se ha diseñado el dispositivo adecuado?
A su vez, las actividades de verificación y validación en el proceso de diseño de dispositivos médicos / producto sanitario, deben estar respaldadas por un sólido plan de gestión de riesgos, en línea con las normas ISO 14971, y cumplir con los procedimientos del sistema de gestión de calidad correspondiente (alineado con ISO 13485).
Es importante tener en consideración que dentro del ejercicio de gestión de riesgos se haya identificado el diseño más crítico o el peor caso, considerando los riesgos identificados y los resultados. Puede ser necesario evaluar varios diseños críticos para demostrar un control efectivo de los riesgos asociados.
El nivel de formalidad de las pruebas será determinado a su vez bajo un ejercicio de gestión de riesgos.
Figura 1: Aplicación de controles de diseño al proceso de diseño en cascada (figura utilizada con autorización de la Oficina de Dispositivos Médicos, Health Canada)
En resumen, el propósito principal de las actividades de verificación y validación de diseño es garantizar que el dispositivo se encuentra dentro de los límites del diseño especificado, cumplan con las necesidades del usuario y de su uso previsto, dentro de un proceso controlado y trazable.
Actividades de verificación
Las actividades de verificación se llevan a cabo en todas las etapas y niveles del diseño del dispositivo. Cualquier enfoque que establezca la conformidad con un requisito de entrada del diseño es un medio aceptable para verificar el diseño con respecto a ese requisito. En muchos casos, son posibles una variedad de enfoques. En línea con la IEC 62304, podemos distinguir diferentes niveles de verificación:
Ensayos de la unidad del software. Ensayos dirigidos a verificar la implementación del diseño para cada elemento de software.
Ensayos de integración del software. Ensayos que verifican la completa integración del elemento de software en el sistema software. Se trata de un proceso progresivo hasta la completa integración del sistema software. Incluye los ensayos de regresión que comprueban las capas adicionales de software conforme no introducen errores en el software previamente integrado.
Ensayos del sistema software. Ensayos dirigidos a verificar el sistema de software de forma completa.
Actividades de Validación
Las actividades de validación del diseño prosiguen a las de verificación, una vez éstas han concluido de forma exitosa y ya se dispone del dispositivo acabado o equivalente. Esta actividad puede incluir unidades de producción inicial, lotes o sus equivalentes con la justificación de la elección del producto, y su evaluación clínica.
Los pasos a seguir en el proceso de validación del diseño son los siguientes:
Desarrollo del Plan de validación. Se trata de un documento de sistemática, definición de alcances y estrategia. Este documento debe incluir la identificación del alcance de las actividades de validación, así como su enfoque de especificación/ensayo, restricciones posibles, criterios de aceptación, entornos de trabajo, recursos, cronología, y metodologías a aplicar. Es importante revisar la incorporación al plan del cumplimiento de los requisitos reguladores , así como hacer referencia a otro tipo de actividades relacionadas, como por ejemplo, las de especificación/verificación, o de evaluación clínica.
Desarrollo de un ejercicio de evaluación de riesgos integrado y completo teniendo en cuenta el uso previsto, y los factores de influencia secundarios, como componentes de interfaz del sistema/producto final o el modo de montaje.
Redacción del Plan de pruebas que incluya los criterios de aceptación y los resultados. Deben incluirse los casos de test con los escenarios comunes que demuestren que el software cumple con su uso previsto. Están diseñados para descubrir errores potenciales y demostrar si las funciones clave funcionan correctamente.
Ejecución del Plan de pruebas en arreglo a los procedimientos de prueba, los requisitos reglamentarios, y buenas prácticas documentales.
Informe final de validación con el resumen de resultados e información de soporte para la adecuada liberación del software. Es la respuesta al Plan de Validación. En esta etapa, se aprovecha a su vez para la revisión de los procedimientos de uso / instrucciones, así como el riesgo residual final.
Remarcar que las actividades de verificación y validación no se limitan a una actividad única, ya que debe asegurase que el sistema software es conforme tras los cambios de configuración o mejoras que puedan llegar a producirse a lo largo de su ciclo de vida.
Figura 2: Ciclo de vida del software
Trazabilidad y automatización
Tanto para las actividades de Verificación, así como de validación, es muy importante contar con una Matriz de Trazabilidad de Requisitos (RTM) que asegure la correcta trazabilidad entre requisitos y ensayos de forma sencilla y eficaz.
A su vez, las nuevas herramientas existentes en el mercado para la automatización de las actividades a lo largo del desarrollo de software, así como de validación digital, hacen posible una mejor gestión de las actividades, permitiendo la trazabilidad de cada requisito, con su especificación, análisis, y ensayo/test de verificación y validación, incluyendo las evidencias, así como una reducción de los errores humanos.
Este enfoque para la mejora de la trazabilidad es especialmente útil para detectar suposiciones o premisas ocultas. Las suposiciones ocultas son peligrosas porque a menudo conducen a un diseño excesivo, lo que añade costes y complejidad innecesarios al diseño. En otros casos, resultan en un diseño no documentado.
Ciberseguridad
Las actuales regulaciones sobre dispositivos médicos están evolucionando para garantizar que los dispositivos comercializados sean aptos para los nuevos desafíos tecnológicos relacionados con los riesgos en materia de ciberseguridad.
En la actualidad, existen numerosos documentos guía publicados por los diferentes organismos reguladores que debemos tener en cuenta a la hora de implementar las medidas de ciberseguridad más adecuadas en nuestros dispositivos.
En especial debemos prestar atención a los requisitos esenciales de seguridad para todos los dispositivos médicos que incorporen sistemas electrónicos programables (PEMS, por sus siglas en inglés) y software que sean en sí mismos dispositivos médicos. Debemos aplicar en este campo principios de gestión de riesgos, incluida la seguridad de la información, así como establecer requisitos mínimos de seguridad informática, incluida la protección contra el acceso no autorizado.
Por otro lado, existen muchos dispositivos que no fueron diseñados con estas mismas consideraciones y exigencias, por lo que pueden presentar riesgos para los pacientes. Por ello, debemos abordar sus amenazas de ciberseguridad, tal y como recomiendan las regulaciones y buenas prácticas actuales.
Pensamiento crítico y enfoque CSA
El enfoque CSA de la CDRH (FDA) intenta clarificar la interpretación de los requisitos regulatorios con el entorno tecnológico actual, para cambiar el foco de la documentación al pensamiento crítico durante todo el ciclo de vida del software de un dispositivo médico.
La “garantía de la calidad del software” se debe centrar en prevenir la introducción de defectos en el proceso de desarrollo de software y fomenta el uso de un enfoque basado en el riesgo para establecer la confianza conforme el software es adecuado para su uso previsto.
Este nuevo enfoque promueve por lo tanto buenas prácticas de calidad orientadas en los siguientes conceptos:
Aprovechar la experiencia de los expertos en la materia (SME) aplicando el pensamiento crítico para determinar la metodología y tecnología de verificación más apropiada basada en el riesgo.
Generación de evidencias documentales de soporte siempre y cuando aporten un valor añadido a la calidad de las pruebas.
Mayor dedicación a la verificación activa para la búsqueda de defectos y menor dedicación a la generación de especificaciones por adelantado.
Cambio de foco desde la documentación al pensamiento crítico y a la verificación.
Figura 3: Enfoque CSA y pensamiento crítico
Machine Learning y Artificial Intelligence
En la actualidad se ha acelerado la adopción y el uso de tecnología AI/ML en dispositivos médicos. Este tipo de dispositivos tienen el potencial de transformar la atención médica al obtener conocimientos nuevos y relevantes en consecuencia de la gran cantidad de datos generados durante todas las fases del proceso de atención médica.
Uno de los mayores beneficios de este tipo de dispositivos reside en la oportunidad de seguir aprendiendo e iterando a medida que hay datos adicionales disponibles, incluso del uso y la experiencia en el mundo real, para mejorar su rendimiento.
Al ser aún una tecnología con una introducción aún incipiente, las entidades reguladoras están publicando documentos de reflexión y concepto sobre la materia para alertar a la industria sobre sus riesgos, para la no introducción de sesgos, clarificación de responsabilidades, mantenimiento de la privacidad, seguridad y confidencialidad de los datos, así como de no introducción de errores por una no adecuada fiabilidad y precisión de los resultados.
Normativas de referencia
Las empresas fabricantes de software de dispositivos médicos deben disponer de un sistema de gestión de calidad adecuado que asegure el correcto ciclo de vida del dispositivo médico. A continuación, se dan como referencia y orientación las siguientes guías y estándares:
Estándares
ISO 13485 Medical Devices – Quality Management Systems – Requirements for Regulatory Purposes
ISO 14971 Medical Devices – Application of Risk Management to Medical Devices
ISO 14155 Clinical Investigation of Medical Devices for Human Subjects: Good Clinical Practice
IEC 62366-1 Medical devices – Part 1: Application of usability engineering to medical devices
IEC 62304:2006 Medical device software. Software life cycle processes.
IEC 82304-1:2016 Health software – Part 1: General requirements for product safety
ISO/TS 82304-2:2021 Health software – Part 2: Health and wellness apps. Quality and reliability
IEC 81001-5-1 IEC 81001-5-1 Health software and health IT systems safety, effectiveness and security.
ISO/IEC TR 24028:2020 Information technology — Artificial intelligence — Overview of trustworthiness in artificial intelligence
ISO/IEC DIS 5259-1 Artificial intelligence — Data quality for analytics and machine learning (ML) Under development (draft 2023)
Documentos guías publicados por Autoridades Regulatorias
CDRH. Draft Guidance – Computer Software Assurance for Production and Quality System Software. Sep 2022.
Europe MDCG 2021-3, Questions and Answers on Custom-Made Devices (& considerations on Adaptable medical devices and Patient-matched medical devices). Mar 2021.
US FDA 21 CFR 820.30, Design Control Guidance for Medical Device Manufacturers. Mar 1997.
US FDA CDRH, Technical Considerations for Additive Manufactured Devices – Guidance for Industry and Food and Drug Administration Staff. Dec 2017.
US FDA CDRH, Applying Human Factors and Usability Engineering to Medical Devices – Guidance for Industry and Food and Drug Administration Staff. Feb 2016.
US FDA CDER, CBER – Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients – Guidance for Industry (Revision 1). Sept 2016.
MITRE_MDIC (FDA) – Playbook for Threat Modeling Medical Devices.
Cybersecurity for Networked Medical Devices Containing Off-the-Shelf (OTS) Software.
MDCG 2019-16—Guidance on Cybersecurity for Medical Devices.
Australia TGA, Guidance on Personalized Medical Devices (including 3D-printed Devices) regulatory reforms. 2022.
Health Canada, Supporting Evidence for Implantable Medical Devices Manufactured by 3D Printing. Apr 2019.
China NMPA, Technical Review Guidance for the Registration of Personalized Additive Manufacturing Medical Devices of Passive Implantable Bone, Joint and Oral Hard Tissues.
Japan MHLW, Guidance on Evaluation of Customized Orthopedic Devices for Osteosynthesis. Dec 2010.
Japan MHLW, Guidance on Evaluation of Orthopedic Customized Artificial Hip Joint Prosthesis. Dec 2011.
Singapore HSA, Regulatory Guideline for 3D-Printed Medical Devices, July 2021
South Korea MFDS, Guidance for Patient-matched Medical Devices manufactured using 3D printers. Dec 2015.
Discussion Papers. AI in Drug Manufacturing, (March 2023);
Using Artificial Intelligence and ML in the Development of Drug and Biological Products. FDA, CDER. May 2023.
Reflection paper on the use of Artificial Intelligence (AI) in the medicinal product lifecycle. EMA, CVMP & CHMP. Jul 2023.
GMLP for Medical Device Development: Guiding Principles. MHRA, Health Canada, FDA. Oct 2021.
Documentos guías publicados por IMDRF / GHTF
GHTF/SG3/N99-10:2004 (Edition 2) Quality Management Systems – Process Validation Guidance.
GHTF/SG1/N71:2012 Definitions of the Terms ‘Medical Device’ and ‘In Vitro Diagnostic (IVD) Medical Device’.
GHTF/SC/N4:2012 (Edition 2) Glossary and definition of terms used in GHTF documents.
IMDRF Principles and Practices for Medical Device Cybersecurity.
Documentos guías publicados por la industria
ISPE. GAMP 5 Guide 2nd Edition. A Risk-Based Approach to Compliant GxP Computerized Systems. July 2022.
ISPE. GAMP Good Practice Guide: Enabling Innovation. Sep 2021.
ISPE. RDI GAMP Guides: Records and data integrity.
AAMI TIR45:2023; Guidance on the use of agile practices in the development of medical device software.
¿Qué ofrece Trescal?
Desde TRESCAL ofrecemos servicios de soporte relativos al software de dispositivo médico según las normativas de aplicación, ofreciendo consultoría de calidad específica en las siguientes materias:
Auditoría de software.
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Proceso de desarrollo y mantenimiento del software de dispositivo médico (IEC 62304).
Desarrollo del VMP / VP.
Establecimiento de estrategias.
Desarrollo de los expedientes de validación.
Actividades de verificación y validación.
Transición a enfoque CSA (Computer Software Assurance).
Cualificacion de Infraestructura IT
Evaluación de proveedores de SW / tecnológicos.
Acompañamiento a proveedores en pruebas de aceptación.
Procedimientos del sistema de gestión de calidad.
Actividades de formación dirigida.
TRESCAL es un proveedor global de servicios para la industria LIfe Sciences que ofrece servicios de implantación/consultoría en sistemas de calidad de productos sanitarios, gestión de riesgos, Data Integrity, validación de sistemas, cualificación de equipos, calibraciones (incluyendo calibraciones dentro del sector médico/sanitario). Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del proyecto de validación, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Acrónimos
AI: Artificial Intelligence
CDRH: Center for Devices and Radiological Health
CSA: Computer Software Assurance
FDA: Food and Drug Administration
GHTF: Global Harmonization Task Force
IMDRF: international Medical Device Regulators Forum
ML: Machine Learning
PMS: Programmable Electrical Medical Systems
RTM: Requirements Traceability Matrix
SME: Subject-Matter Expert
SW: Software
Imagen1
SW de dispositivos médicos / producto sanitario: ciclo de vida y validación. Parte 1
Introducción
La rápida evolución en la tecnología existente está revolucionando la forma en la que nos comunicamos y gestionamos la información. La digitalización de los dispositivos médicos para dotarlos de tecnologías avanzadas clave de la industria 4.0. no es un asunto desconocido dentro del sector de dispositivos médicos, en donde el uso de dispositivos médicos inteligentes es ya una clara apuesta y tendencia del sector. Se estima que uno de cada cuatro dispositivos médicos incorpora software para dispositivos médicos o son dispositivos médicos por sí mismos.
Por otro lado, la digitalización de la atención sanitaria está permitiendo a pacientes y médicos interactuar con la información de salud de una manera sin precedentes para su mejor optimización y sostenibilidad. Como ejemplos clave se encuentran la tecnología Cloud, Big Data, IoT o AI/ML.
A su vez, este proceso de digitalización está generando una enorme cantidad de datos que supera con creces las capacidades analíticas de los médicos a nivel individual, por lo que estamos viviendo un gran auge de herramientas de inteligencia artificial como catalizador del análisis de datos. Además, no debemos olvidarnos de la gran eclosión del uso generalizado de teléfonos inteligentes y productos digitales portátiles.
El software se ha convertido en consecuencia en una parte importante del panorama del campo de los dispositivos médicos o productos sanitarios, y debemos implementarlo de una forma segura y efectiva para asegurar el uso y seguridad del dispositivo médico en cuestión.
Definiciones de SW relacionado con dispositivos médicos o productos sanitarios
A nivel mundial, existen muchos mercados para la comercialización de dispositivos médicos, pero la Unión Europea y Estados Unidos son quizás los de mayor relevancia al ser los más grandes.
Es importante introducir y remarcar que las regulaciones de la Unión Europea y de los Estados Unidos no utilizan los mismos términos en referencia al SW relacionado con dispositivos médicos. Por un lado, la Unión Europea habla de MDSW (Medical Device Software), y Estados Unidos de SaMD (Software as a Medical Device).
Aclarar que el término SaMD es un concepto internacional, según lo define el International Medical Device Regulators Forum (IMDRF), y que éste se incluye en la legislación de muchos países (a parte de la de Estados Unidos, como es por ejemplo la del Reino Unido, o la del Canadá), mientras que MDSW es exclusivamente un término utilizado por la Unión Europea.
A continuación, revisamos y resumimos cada enfoque, y los matices más relevantes a tener en cuenta:
Estados Unidos: SaMD, SiMD
Para la FDA el software relacionado con los dispositivos médicos puede ser catalogado dentro de las siguientes tipologías:
Software como Dispositivo Médico (SaMD): Software que por sí solo es un dispositivo médico. Es decir, es aquel que «está destinado a ser utilizado para uno o más fines médicos sin ser parte de un dispositivo de hardware«. La FDA define SaMD utilizando la descripción presentada por el International Medical Device Regulators Forum (IMDRF), de la que es miembro. Para ello, debemos conocer bien el uso previsto del producto y sus indicaciones de uso, y estar en conformidad con la definición de dispositivo de la 181 sección 201(h) de la FD&C Act.
Software en un Dispositivo Médico (SiMD): Software que es parte integral de un dispositivo médico que es utilizado para operar o controlar un dispositivo de hardware pero que no cumple con los requisitos para ser categorizado como SaMD. Es decir, es aquel que ayuda a ejecutar un dispositivo médico de hardware (por ejemplo, alimentando su mecánica o produciendo una interfaz gráfica). Sin este software, el dispositivo no podría funcionar correctamente. Un “software embebido” o “firmware” que forma parte de un dispositivo médico de hardware, o un accesorio SW para el control remoto o pantalla del dispositivo médico hardware no cumpliría con la definición de SaMD.
Software utilizado en la fabricación o mantenimiento de un dispositivo médico.
A continuación, revisamos unos ejemplos de cuándo un software es SaMD y no lo es:
Software como Dispositivo Médico (SaMD)
Software que proporciona información (input) a otro dispositivo médico con hardware o software médico diferente. Por ejemplo, el software de planificación de tratamientos que suministra información utilizada en un acelerador lineal.
El software con uso previsto médico que opera en una plataforma hardware convencional (ej.: ordenador, tableta, móvil). Por ejemplo, el software destinado al diagnóstico de una enfermedad.
El software que está conectado a un dispositivo médico que contiene hardware pero que no es necesario para que ese producto alcance su uso previsto.
Software que no es un Dispositivo Médico (SaMD)
Software utilizado para «accionar o controlar» los motores y la bomba de infusión de medicamentos, o software que controla un marcapasos (SiMD).
Software requerido por un dispositivo médico de hardware para realizar el uso previsto del dispositivo médico de hardware, incluso si se vende por separado del dispositivo médico de hardware (SiMD).
Software que se basa en datos de un dispositivo médico, pero que no tiene un propósito médico, por ejemplo, software que cifra datos para su transmisión desde un dispositivo médico.
Software que permite la comunicación clínica y el flujo de trabajo, incluido el registro de pacientes, la programación de visitas, las llamadas de voz y las videollamadas.
Software que monitoriza el rendimiento o el funcionamiento adecuado de un dispositivo con el fin de darle servicio (por ejemplo, software que monitoriza el rendimiento del tubo de rayos X para anticipar la necesidad de reemplazo), o software que integra y analiza datos de control de calidad de laboratorio para identificar errores aleatorios o tendencias en la calibración de los IVDs.
Software que proporciona parámetros que se convierten en entradas (inputs) para el software como dispositivo médico pero que no tiene un propósito médico. Por ejemplo, una base de datos que incluye funciones de búsqueda y consulta por sí misma.
En el ámbito de la Unión Europea el software de dispositivo médico (MDSW) es el software destinado a ser utilizado, solo o en combinación, para un propósito especificado en la definición de «dispositivo médico» conforme a la MDR o IVDR, independientemente que el software sea independiente o impulse o influya en el uso de un dispositivo.
En resumen, se deben cumplir las siguientes tres condiciones para que el software sea clasificado como MDSW:
Tener un propósito médico por sí solo,
Realizar una acción sobre datos más allá del almacenamiento, archivo, comunicación, búsqueda simple o compresión sin pérdidas, y
Estar destinado al beneficio de pacientes individuales.
Cabe mencionar que los siguientes tipos de software que no tienen un propósito de dispositivo médico en sí mismos y, por lo tanto, no se consideran MDSW, aún están sujetos a los requisitos del EU MDR o IVDR, según corresponda:
Software que corresponde a la definición legal de «accesorio» de un dispositivo médico o de un dispositivo de diagnóstico in vitro (IVD),
Software que corresponde a productos sin fines médicos, según lo enumerado en el Anexo XVI del MDR de la EU,
Software que impulsa o influye en el uso de un dispositivo médico (hardware) o IVD, siempre que no tenga un propósito médico separado y no cree información por sí solo (en cuyo caso, el módulo de software con un propósito médico separado o que genere información cualificaría como MDSW). Este software puede, entre otros ejemplos:
(a) operar, modificar el estado o controlar el dispositivo ya sea a través de una interfaz (por ejemplo, software, hardware) o a través del operador de este dispositivo
(b) o suministrar salida relacionada con el funcionamiento (hardware) de ese dispositivo
MDCG 2023-4: Medical Device Software (MDSW) – Hardware combinations Guidance on MDSW intended to work in combination with hardware or hardware components
Manual: Manual on borderline and classification under Regulations (EU) 2017/745 and 2017/746
Infografía: Is your software a Medical Device?
MDCG 2021-24: Guidance on classification of medical devices
MDCG 2020-1: Guidance on clinical evaluation (MDR) / Performance evaluation (IVDR) of medical device software.
MDCG 2019-16: Guidance on cybersecurity for medical devices
MDCG 2019-11: Qualification and classification of software – Regulation (EU) 2017/745 and Regulation (EU) 2017/746.
Diferencias entre MDSW y SaMD
Mientras que SaMD se refiere únicamente al software que es independiente y excluye completamente a cualquier software “embebido” en un dispositivo médico físico, MDSW puede ser independiente o parte de un dispositivo médico de hardware, en el caso de que tenga su propio propósito de dispositivo médico además del propósito de impulsar/influir en el dispositivo médico de hardware. En MDCG 2019-11 se proporcionan un par de ejemplos de software “embebido” con fines médicos propios:
Software de análisis de imágenes de melanoma destinado a controlar un escáner de luz láser de infrarrojo cercano.
MDSW pretende medir y transmitir los niveles de glucosa en sangre, calcular la dosis de insulina necesaria.
Por otro lado, el término de software “standalone” (o independiente) utilizado ampliamente en la industria de la tecnología médica no es sinónimo de MDSW «standalone» ya que un MDSW «standalone» aún puede controlar o influir en un dispositivo médico de hardware.
Ciclo de vida del SW de dispositivos médicos: IEC 62304
La IEC 62304 es una norma internacional que establece requisitos y directrices para el ciclo de vida del software de dispositivos médicos. Esta norma es un estándar de consenso reconocido por las agencias de salud que establece la seguridad y eficacia de un dispositivo médico que contiene software.
El objetivo principal de la IEC 62304 es proporcionar un marco de trabajo para el desarrollo y mantenimiento de software en dispositivos médicos, asegurando la seguridad y eficacia de estos productos. La norma se enfoca en la gestión de riesgos, la documentación, la validación y verificación del software, y garantiza que se cumplan los requisitos reglamentarios y de calidad propios de la industria de dispositivos médicos.
Por otro lado, la IEC 62304 no cubre la parte de validación clínica del sistema SW como dispositivo médico, sino la parte técnica, no clínica, desde la liberación hasta el mantenimiento y discontinuación.
Según este estándar los procesos del ciclo de vida del software de dispositivo médico son los siguientes:
Desarrollo del software.
Mantenimiento del software.
Gestión de riesgos del software.
Gestión de la configuración del software.
Resolución de problemas del software.
Desarrollo del software
El proceso de desarrollo del software está comprendido por las siguientes etapas:
Plan de desarrollo del software
Análisis de requisitos del software.
Diseño arquitectónico del software.
Diseño detallado del software.
Implementación y verificación de la unidad software.
Integración y pruebas del sistema de software.
Liberación de software.
Pero antes de iniciar el proceso de desarrollo del SW, los fabricantes de dispositivos médicos deben realizar una clasificación de la seguridad del software para la asignación de una clase (A, B o C) a cada sistema de software que se implemente en el dispositivo médico según su severidad conforme a los posibles efectos adversos que puedan causar sobre el paciente. Esta severidad puede ser la siguiente:
Clase A: no es posible que se produzcan lesiones ni daños a la salud.
Clase B: es posible que se produzcan lesiones o daños no graves a la salud.
Clase C: es posible la muerte o lesiones graves.
El propósito de realizar esta clasificación de seguridad del software sirve de base para determinar qué tan riguroso debe ser el proceso de desarrollo de software posterior (ver figura 1):
Capítulo
Clase A
Clase B
Clase C
Planificación del desarrollo (5.1.)
x
x
x
Análisis de los requisitos (5.2)
x
x
x
Diseño arquitectura (5.3)
x
x
Diseño detallado (5.4)
x
x
Implementación (5.5)
x
x
x
Ensayos test unitarios (5.5)
x
x
Integración (5.6)
x
x
Ensayos de integración (5.6)
x
x
Ensayos del sistema (5.7)
x
x
x
Liberación (5.8)
x
x
x
Figura 1: Actividades recogidas en 62304 en función del riesgo del dispositivo médico
Por otro lado, tener en cuenta que aunque la IEC 62304 tiene una estructura bastante lineal es posible a su vez cumplir con sus requisitos utilizando un desarrollo de SW ágil. De hecho, existe otro estándar AAMI TIR 45 que ofrece orientación sobre el uso de prácticas ágiles en el desarrollo de software de dispositivos médicos.
Es por ello importante la definición de un modelo de ciclo de desarrollo del software acorde a los estándares actuales que pueda tener cabida a enfoques agiles de desarrollo de SW más flexibles, dinámicos e incrementales, especialmente ante sistemas de software complejos en donde se necesita un mayor ajuste y redefinición de requisitos.
Mantenimiento del software
Debe establecerse un plan de mantenimiento que describe cómo se gestionará el mantenimiento del software a lo largo de su ciclo de vida. Esto incluye la identificación de las actividades de mantenimiento necesarias, la frecuencia de las actualizaciones, la gestión de riesgos asociados con el mantenimiento, y la asignación de recursos y responsabilidades.
El proceso de mantenimiento del software descrito en IEC 62304 consta de los siguientes puntos:
Establecimiento del plan de mantenimiento del software
Análisis de problemas y modificaciones.
Implementación de las modificaciones.
Gestión de riesgos del software
Los requisitos de gestión de riesgos de la IEC 62304 se correlacionan con los de ISO 14971, norma internacional sobre la aplicación de la gestión de riesgos en dispositivos médicos, por lo que resulta fácil notar su interconexión. Este proceso implica la identificación, análisis, evaluación y gestión de los riesgos asociados con el software médico a lo largo de todo su ciclo de vida. Esto incluye, por ejemplo:
Análisis del software en términos de contribución a situaciones peligrosas.
Medidas de control de riesgo.
Verificación de las medidas de control de riesgo.
Gestión de riesgos de cambios en el software.
Gestión de la configuración del software
Este proceso se encarga de mantener un control estricto de las configuraciones y versiones del software a lo largo de su ciclo de vida permitiendo su trazabilidad. De esta forma se identifican y definen los elementos de software (incluyendo la documentación), así como se controlan los cambios y liberaciones, documentando e informando del estado de los elementos de configuración y solicitudes de cambio. Se incluye:
Identificación de la configuración: Identificación de cada elemento de configuración del software y sus versiones.
Control de Cambios: Incluyendo el proceso de solicitud de cambios.
Histórico del control de los elementos de configuración.
Resolución de problemas del software
Finalmente, el proceso de resolución de problemas del software implica la identificación y gestión de problemas, errores o no conformidades en el software, así como la implementación de acciones correctivas para su resolución. Este proceso proporciona un medio oportuno, responsable y documentado para garantizar que el problema es analizado y resuelto.
IEC 62304 establece un esquema general para un proceso de resolución de problemas:
Elaboración de los informes de problemas
Estudio del problema
Aviso a las partes interesadas
Uso del proceso de control de cambios
Mantenimiento de los registros
Análisis para la detección de tendencias en los informes de problemas
Verificación de la resolución de problemas de software
Contenido de la documentación de prueba/ensayo
Como regla general, es probable que encuentre menos problemas con el tiempo a medida que su producto madure. Sin embargo, es probable que la gravedad de los problemas que encuentre más adelante sea aún mayor y requiera un proceso más riguroso para resolución.
Normativas de referencia
Las empresas fabricantes de software de dispositivos médicos deben disponer de un sistema de gestión de calidad adecuado que asegure el correcto ciclo de vida del dispositivo médico. A continuación, se dan como referencia y orientación las siguientes guías y estándares:
Estándares
ISO 13485 Medical Devices – Quality Management Systems – Requirements for Regulatory Purposes
ISO 14971 Medical Devices – Application of Risk Management to Medical Devices
ISO 14155 Clinical Investigation of Medical Devices for Human Subjects: Good Clinical Practice
IEC 62366-1 Medical devices – Part 1: Application of usability engineering to medical devices
IEC 62304:2006 Medical device software. Software life cycle processes.
IEC 82304-1:2016 Health software – Part 1: General requirements for product safety
ISO/TS 82304-2:2021 Health software – Part 2: Health and wellness apps. Quality and reliability
IEC 81001-5-1 IEC 81001-5-1 Health software and health IT systems safety, effectiveness and security.
ISO/IEC TR 24028:2020 Information technology — Artificial intelligence — Overview of trustworthiness in artificial intelligence
ISO/IEC DIS 5259-1 Artificial intelligence — Data quality for analytics and machine learning (ML) Under development (draft 2023)
Documentos guías publicados por Autoridades Regulatorias
CDRH. Draft Guidance – Computer Software Assurance for Production and Quality System Software. Sep 2022.
Europe MDCG 2021-3, Questions and Answers on Custom-Made Devices (& considerations on Adaptable medical devices and Patient-matched medical devices). Mar 2021.
US FDA 21 CFR 820.30, Design Control Guidance for Medical Device Manufacturers. Mar 1997.
US FDA CDRH, Technical Considerations for Additive Manufactured Devices – Guidance for Industry and Food and Drug Administration Staff. Dec 2017.
US FDA CDRH, Applying Human Factors and Usability Engineering to Medical Devices – Guidance for Industry and Food and Drug Administration Staff. Feb 2016.
US FDA CDER, CBER – Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients – Guidance for Industry (Revision 1). Sept 2016.
MITRE_MDIC (FDA) – Playbook for Threat Modeling Medical Devices.
Cybersecurity for Networked Medical Devices Containing Off-the-Shelf (OTS) Software.
MDCG 2019-16—Guidance on Cybersecurity for Medical Devices.
Australia TGA, Guidance on Personalized Medical Devices (including 3D-printed Devices) regulatory reforms. 2022.
Health Canada, Supporting Evidence for Implantable Medical Devices Manufactured by 3D Printing. Apr 2019.
China NMPA, Technical Review Guidance for the Registration of Personalized Additive Manufacturing Medical Devices of Passive Implantable Bone, Joint and Oral Hard Tissues.
Japan MHLW, Guidance on Evaluation of Customized Orthopedic Devices for Osteosynthesis. Dec 2010.
Japan MHLW, Guidance on Evaluation of Orthopedic Customized Artificial Hip Joint Prosthesis. Dec 2011.
Singapore HSA, Regulatory Guideline for 3D-Printed Medical Devices, July 2021
South Korea MFDS, Guidance for Patient-matched Medical Devices manufactured using 3D printers. Dec 2015.
Discussion Papers. AI in Drug Manufacturing, (March 2023);
Using Artificial Intelligence and ML in the Development of Drug and Biological Products. FDA, CDER. May 2023.
Reflection paper on the use of Artificial Intelligence (AI) in the medicinal product lifecycle. EMA, CVMP & CHMP. Jul 2023.
GMLP for Medical Device Development: Guiding Principles. MHRA, Health Canada, FDA. Oct 2021.
Documentos guías publicados por IMDRF / GHTF
GHTF/SG3/N99-10:2004 (Edition 2) Quality Management Systems – Process Validation Guidance.
GHTF/SG1/N71:2012 Definitions of the Terms ‘Medical Device’ and ‘In Vitro Diagnostic (IVD) Medical Device’.
GHTF/SC/N4:2012 (Edition 2) Glossary and definition of terms used in GHTF documents.
IMDRF Principles and Practices for Medical Device Cybersecurity.
Documentos guías publicados por la industria
ISPE. GAMP 5 Guide 2nd Edition. A Risk-Based Approach to Compliant GxP Computerized Systems. July 2022.
ISPE. GAMP Good Practice Guide: Enabling Innovation. Sep 2021.
ISPE. RDI GAMP Guides: Records and data integrity.
AAMI TIR45:2023; Guidance on the use of agile practices in the development of medical device software.
¿Qué ofrece Trescal?nbsp;
Desde TRESCAL ofrecemos servicios de soporte relativos al software de dispositivo médico según las normativas de aplicación, ofreciendo consultoría de calidad específica en las siguientes materias:
Auditoría de software.
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Proceso de desarrollo y mantenimiento del software de dispositivo médico (IEC 62304).
Desarrollo del VMP / VP.
Establecimiento de estrategias.
Desarrollo de los expedientes de validación.
Actividades de verificación y validación.
Transición a enfoque CSA (Computer Software Assurance).
Cualificacion de Infraestructura IT
Evaluación de proveedores de SW / tecnológicos.
Acompañamiento a proveedores en pruebas de aceptación.
Procedimientos del sistema de gestión de calidad.
Actividades de formación dirigida.
TRESCAL es un proveedor global de servicios para la industria LIfe Sciences que ofrece servicios de implantación/consultoría en sistemas de calidad de productos sanitarios, gestión de riesgos, Data Integrity, validación de sistemas, cualificación de equipos, calibraciones (incluyendo calibraciones dentro del sector médico/sanitario). Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del proyecto de validación, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Acrónimos
AI: Artificial Intelligence
EC: European Comission
EU: European Union
FDA: Food and Drug Administration
GHTF: Global Harmonization Task Force
IMDRF: international Medical Device Regulators Forum
IoT: Internet of Things
IVD: in vitro diagnostic medical device
IVDR: In Vitro Medical Device Regulation (EU) 2017/746
MDCG: Medical Device Coordination Group
MDR: Medical Device Regulation (EU) 2017/745
MDSW: Medical Device Software
ML: Machine Learning
SaMD: Software as a Medical Device
SiMD: Software in Medical Device
SW: Software
GMP Qualification and validation
PUNTOS CLAVE PARA AFRONTAR EL NUEVO ANEXO 1 EU GMP
El nuevo anexo 1 representa un reto y un buen recordatorio para los fabricantes de productos estériles (y no estériles) en la mejora del sistema PQS (Pharmaceutical Quality System), para su mejor alineación en materia de control y prevención de la contaminación del producto final. Como cualquier otro proyecto de mejora, debemos planificar y estructurar las diferentes actividades a llevar a cabo para la consecución de los nuevos objetivos de forma óptima y efectiva, en búsqueda de una mejor conformidad de las instalaciones y procesos de fabricación.
Para llevar a cabo esta andadura con éxito es primordial tener en cuenta los siguientes aspectos:
Creación de un equipo multifuncional experto en la materia. Debe existir un adecuado trabajo de equipo y una buena comunicación desde el inicio del proyecto entre todas las partes y departamentos involucrados (ej.: departamento de microbiología, producción, garantía de calidad, control de calidad, validaciones, regulación, mantenimiento), incluyendo expertos en ciencias de los datos, gestión de los riesgos, y proveedores de servicios críticos. En este punto, debemos revisar bien nuestros procesos para delimitar alcances iniciales y responsabilidades.
Conocer el nivel de madurez en materia de control de contaminación, es decir conocer si disponemos de experiencia amplia en fabricación de medicamentos estériles, u otros productos en donde el control de la contaminación es un punto importante.
Recopilación de datos relevantes. Debe realizarse un esfuerzo colectivo de gestión del conocimiento para mejorar y actualizar el conocimiento que se encuentra en documentos y bases de datos. Para ello, recopilaremos toda la información técnica del producto y del proceso, así como nos rodearemos de toda la documentación interna en materia de control de contaminación.
Realizar un análisis de brechas de los cambios requeridos del Anexo 1. En el caso de instalaciones ya existentes, el realizar un Gap Assessment para la identificación de brechas en las novedades del anexo 1, es un ejercicio altamente recomendable para saber a priori qué nivel de brecha tenemos entre la situación actual y la definida por el anexo 1.
Prestar atención al diseño y control de las instalaciones, equipos, servicios y procesos. La evaluación del diseño de la instalación y de los procesos (e incorporación de nuevas tecnologías de fabricación avanzadas, sistemas de barrera -RABS, aisladores-, y nuevas metodologías de monitorización/control), así como la revisión del estado de su estado de cualificación / validación, es fundamental para garantizar que las prácticas actuales cumplen con los nuevos requisitos.
Prestar atención al personal. Los procesos y prácticas en materia de prevención y control de la contaminación pueden requerir cambios dependiendo de la interpretación e implementación previa de los requisitos del Anexo 1. Debe existir formación específica en materia de contaminación (ej.: prácticas de vestimenta, higiene, trabajo aséptico, desinfección y limpieza), así como requerimientos de cualificación y descualificación del personal.
Trabajar con proveedores expertos. Rodearnos de proveedores expertos nos permite una mayor garantía de todos los puntos del proceso de fabricación y cadena de suministro. Su evaluación, así como la elaboración de acuerdos de calidad específicos, son asuntos clave a tener en consideración.
PQS efectivo y cierre de la política CCS Contamination Control Strategy). Debe existir una alineación del CCS con el sistema PQS para la evaluación holística de los riesgos, y medidas de contención existentes. Evaluar el impacto de los cambios del Anexo 1 en los aspectos definidos del CCS debe conllevar una revisión periódica continua del PQS. Es importante el análisis de los datos y tendencias en continuo para el establecimiento de los planes de mejora y mitigación oportunos. La disposición de herramientas de análisis e investigación avanzadas y sólidas juegan aquí un papel relevante.
Sistema QRM (Quality Risk Management) robusto. El contar con un sistema de gestión de los riesgos para la calidad robusto es crucial para el efectivo proceso de toma de decisión basado en los riesgos. Es importante disponer de los datos adecuados, así como elegir la herramienta de gestión de riesgos y el nivel de formalidad del proceso QRM más adecuado en cada etapa del proyecto en base al nivel de nivel de incertidumbre, complejidad, e importancia.
Estrategia de control de contaminación y PQS
Conclusión
Los fabricantes deben adoptar un enfoque basado en el riesgo para analizar sus procesos e instalaciones actuales para la determinación de las medidas necesarias más efectivas para cumplir con los requisitos del nuevo anexo 1 y así mejorar su PQS en términos de control y prevención de la contaminación. Y todo ello, sobre una base de revisión periódica y continua.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, validación de sistemas, cualificación de equipos y calibración. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten:
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Gap análisis de la estrategia actual de control de la contaminación.
Evaluación de proveedores.
Cualificación de salas limpias.
Ensayos de direccionalidad de humo (visualización y filmación)
Cualificación de servicios farmacéuticos.
Cualificación de equipos de esterilización.
Establecimiento y mantenimiento de los planes de verificación de salas limpias.
Desarrollo / revisión de la estrategia de control de la contaminación (CCS).
Validaciones de proceso.
Validaciones de limpieza.
Desarrollo de los expedientes de validación / cualificación.
Acompañamiento a proveedores en pruebas de aceptación.
Revisiones periódicas.
Calibración instrumental.
Actividades de formación dirigida.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar la correcta alineación de su sistema de calidad, procesos e instalaciones con los nuevos requisitos del Anexo 1, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
IMG-20221005-WA0014
TRESCAL PARTICIPARÁ EN FARMAFORUM LOS DÍAS 20 y 21 DE SEPTIEMBRE DE 2023
Madrid, 1 de septiembre, 2023. El grupo Trescal estará presente en Farmaforum la feria de referencia del sector farmacéutico el próximo mes de septiembre.
Con el objetivo de seguir ampliando mercado e impulsar la imagen Trescal en el sector life sciences Trescal participará con el stand F27 en Farmaforum, Madrid. La feria tendrá lugar los días 20 y 21 de septiembre en IFEMA, Pabellón 14.
Un año más una oportunidad única para mantenerse al día sobre las últimas actualidades GxP y de los servicios más actuales.
A continuación, detallamos los talleres que ofrecerán:
20 de septiembre de 12 a 14h: Digitalización, novedades de la guía Gamp5 2nd Edition, y relevancia del proveedor tecnológico
Ponente: Mar Díaz, Responsable Técnica Validaciones
Ubicación: sala talleres Farmaforum
Puedes apuntarte a los mismos mediante el siguiente enlace
20 de septiembre a las 12:45: Mesa Redonda «Del Diseño al éxito»
Ponente: Roberto Español, Responsable Técnico Life Sciences
Formação mínima de 12ºano ou curso profissional com experiência comprovada;
Experiência de 3 anos na atividade de ensaios em Ambientes Limpos;
Valorizamos os conhecimentos de Inglês e/ou Castelhano;
Conhecimentos de MS Office, especialmente em EXCEL;
Carta de condução tipo B;
Flexibilidade horaria e disponibilidade para deslocações nacionais.
REQUISITOS DESEJADOS
Organização e gestão do tempo;
Planificação;
Iniciativa e proatividade;
Facilidade de comunicação.
CAPACIDADES NECESSÁRIAS
Rigor e Sentido crítico;
Trabalho em equipa;
Ágil adaptabilidade.
O QUE OFERECEMOS
Salário adequado à experiência e competência demonstrada;
Seguro de saúde;
Oportunidade de crescimento numa grande multinacional.
AI. Artificial intelligence concept. Abstract wireframe digital human face on streaming matrix digital binary code background. Human head in robot digital computer interpretation. Vector illustration.
Nuevo documento de reflexión de la EMA sobre el uso de AI el en el ciclo de vida del medicamento
La Agencia Europea de Medicamentos (EMA, por sus siglas en inglés) ha publicado recientemente un documento borrador sobre el uso de la inteligencia artificial (AI) en el desarrollo y regulación de los medicamentos de uso humano y veterinarios.
Introducción
Este pasado 19 de julio, la EMA ha publicado un documento borrador (Reflection paper) sobre el uso de la AI en el ciclo de vida del medicamento. El documento está actualmente y hasta el 31 de diciembre, en trámite de consulta pública.
Este documento de reflexión quiere brindar consideraciones sobre el uso de AI (Artificial Intelligence) y ML (Machine Learning) en el ciclo de vida de los medicamentos, desde su descubrimiento, hasta el momento posterior a su autorización. Dado el rápido desarrollo de este campo, el objetivo de este documento es reflexionar sobre los principios científicos que son relevantes para la evaluación regulatoria, cuando estas tecnologías emergentes se aplican para apoyar el desarrollo y uso seguro y eficaz de los medicamentos.
Reconocimiento de nuevas oportunidades y desafíos
El nuevo borrador reconoce que este nuevo campo/tecnología emergente representa una gran oportunidad para la industria para mejorar todas las fases del ciclo de vida del medicamento, pero admite que trae a su vez desafíos, como la comprensión de los algoritmos o los posibles fallos técnicos que pudieran surgir, o el uso de datos de entrenamiento no representativos y equilibrados teniendo en cuenta todas las bases relevantes para la no discriminación de poblaciones (introducción de sesgos), la no protección de datos personales, o la falta de integridad de los datos. Por ello, el borrador de la EMA destaca que el desarrollo de la AI debe estar permanentemente guiado por humanos y cumplir con los requisitos legales y éticos pertinentes.
A su vez, en varios aspectos, como son la gestión y gobernanza de datos, el rigor estadístico, los principios normativos, y las mejores prácticas establecidas son directamente aplicables a la AI/ML, por lo que deben realizarse esfuerzos en todas las organizaciones para integrar la competencia de la ciencia de los datos dentro del desarrollo de medicamentos y la farmacovigilancia.
El objetivo es poder garantizar la seguridad de los pacientes y de los resultados con el fin de promover la confiabilidad de la AI/ML.
En resumen, el uso de AI en el ciclo de vida del medicamento debe estar en cumplimiento con los requisitos legales existentes, considerando la ética y sus principios subyacentes, con el debido respeto a los derechos fundamentales.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de infraestructura IT. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del ciclo de vida de su sistema informático e infraestructura IT, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Este navegador no es compatible con Trescal. Para asegurarse de que puede utilizar todas las funciones, cambie a un navegador web compatible, como Google Chrome, Mozilla Firefox, Microsoft Edge o Safari.
 oppo_0
oppo_0
 TECHNICAL_BNL_ANVERS_2021_PRESSURE (3)
TECHNICAL_BNL_ANVERS_2021_PRESSURE (3) LinkedIn (1)
LinkedIn (1) Imagen1
Imagen1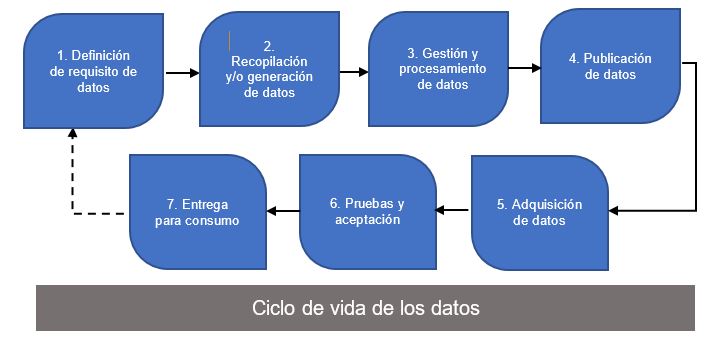

 Imagen1-1
Imagen1-1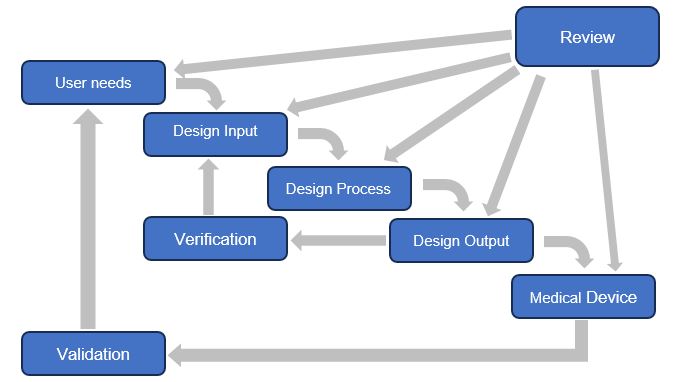
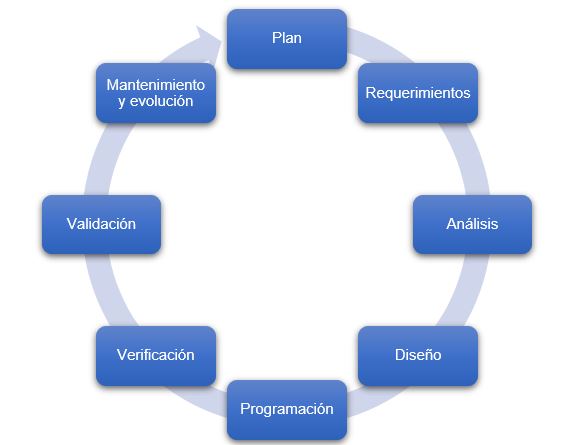
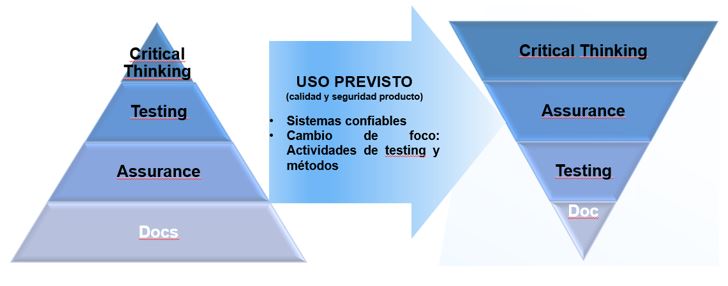
 Imagen1
Imagen1
 GMP Qualification and validation
GMP Qualification and validation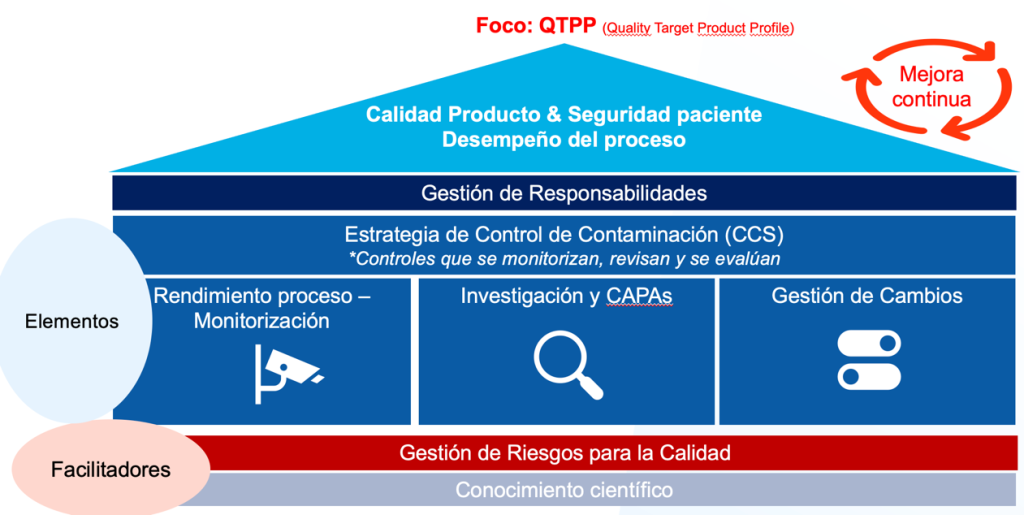
 IMG-20221005-WA0014
IMG-20221005-WA0014 Temperature bath
Temperature bath AI. Artificial intelligence concept. Abstract wireframe digital human face on streaming matrix digital binary code background. Human head in robot digital computer interpretation. Vector illustration.
AI. Artificial intelligence concept. Abstract wireframe digital human face on streaming matrix digital binary code background. Human head in robot digital computer interpretation. Vector illustration.