 Hands placing last piece of a Puzzle
Hands placing last piece of a PuzzleIntroducción y marco previo regulador
Conforme los reglamentos MDR/IVDR, los fabricantes de productos sanitarios deben disponer de un sistema de gestión de calidad, así como un sistema de seguimiento poscomercialización proporcionales a la clase de riesgo y al tipo de producto sanitario en cuestión, a fin de garantizar que los productos fabricados sean conformes. Además, a fin de reducir al mínimo los riesgos y prevenir incidentes relacionados con los productos, los fabricantes deben establecer un sistema para la gestión de riesgos y un sistema para notificar incidentes y acciones correctivas de seguridad.
Dentro de este ámbito, la validación de los sistemas informáticos considerados como críticos para el ciclo de vida del producto es un factor clave a fin de garantizar la calidad y seguridad del producto sanitario.
La ISO 13485:2016 además establece que la organización debe documentar los procedimientos para la validación de los sistemas informáticos utilizados en el sistema de gestión de calidad, y que tales sistemas deben ser validados antes de su uso inicial y, cuando sea apropiado ante cualquier cambio.
Por su parte, la 21 CFR 820 establece la necesidad de validar los sistemas informáticos involucrados en los procesos de la producción o el sistema de calidad. Esto incluye, los procesos relacionados tanto con el diseño de dispositivos médicos, las pruebas de verificación, la aceptación de componentes, la fabricación, el etiquetado, el acondicionado, la distribución, la gestión de reclamaciones o cualquier otro aspecto del sistema de calidad.
Validación de sistemas informáticos
Según la FDA (21 CFR part 11), la validación de un sistema informático es la confirmación mediante examen y provisión de evidencias objetivas que las especificaciones de un sistema informatizado son conformes a los requerimientos del usuario y a su uso previsto, y que todos los requerimientos pueden ser consistentemente cumplidos.
Se trata pues de una actividad destinada a asegurar el uso previsto del sistema informático con el fin de cumplir con el marco regulador aplicable, así como conseguir las autorizaciones y certificaciones necesarias. Esta actividad además nos permite conocer mejor nuestro sistema, sus vulnerabilidades y oportunidades de mejora, no sólo en lo referente a aspectos de calidad, sino también de productividad. Aplicar una buena metodología en nuestro proyecto de validación permite una reducción del coste y tiempo necesarios para conseguir un producto conforme, garantizando el cumplimiento de los requisitos y de las perspectivas reguladoras desde el principio (presentación expediente 510k / marcado de conformidad CE).
Validación del software de dispositivo médico
El software puede ser una parte esencial de un producto sanitario, tanto en el diseño de un producto nuevo, como mejora en la funcionalidad de un dispositivo ya existente. A medida que la tecnología avanza, el número de aplicaciones de software de productos sanitarios también aumenta, y el foco en la seguridad del paciente debe seguir siendo una prioridad de las compañías de dispositivos médicos.
Por otro lado, se siguen notificando problemas sobre falta de implementación de controles de software o en el desarrollo de procedimientos de verificación del software del dispositivo médico que causan la necesidad de corrección o incluso la discontinuidad del producto sanitario. La validación del programa informático dentro del proceso de desarrollo del dispositivo médico es por lo tanto un factor relevante para evitar problemas con la seguridad del producto que impacten en el paciente, así como en la reputación de la empresa.
UNE – EN 62304. Software de dispositivos médicos: Procesos del ciclo de vida del SW
Esta norma aplica al desarrollo y al mantenimiento del software de dispositivos médicos cuando el software es por sí mismo un dispositivo médico o cuando el software es una parte integrante o embebida del dispositivo médico final, y sirve como referente para todo el ciclo de vida del software de Productos Sanitarios (planificación, análisis, especificación y diseño, implementación e integración, verificación, mantenimiento).
Según esta norma, los dispositivos médicos se pueden clasificar en tres categorías según el riesgo de daño en el paciente, operador u otras personas resultantes de una situación peligrosa a la que el sistema software pudiera ocasionar, siendo éstas las siguientes:
- Clase A: El sistema software no puede contribuir a una situación peligrosa, o puede contribuir a una situación peligrosa que no resulte en un riesgo inaceptable tras considerar las medidas externas de control de riesgos aplicadas al sistema software.
- Clase B: El sistema software puede contribuir a una situación peligrosa que resulte en un riesgo inaceptable tras considerar las medidas externas de control de riesgos aplicadas al sistema software y el posible daño resultante es una lesión no seria.
- Clase C: El sistema software puede contribuir a una situación peligrosa que resulte en un riesgo inaceptable tras considerar las medidas externas de control de riesgos aplicadas al sistema software y el posible daño resultante es una lesión seria.
A su vez, el proceso de desarrollo del software de dispositivo médico debe cubrir las siguientes etapas:
- Plan de desarrollo del software
- Análisis de requisitos del software.
- Diseño arquitectónico del software.
- Diseño detallado del software.
- Implementación y verificación de la unidad software.
- Integración y pruebas del sistema de software.
- Liberación de software.
Es importante por ello, la definición de un modelo de ciclo de vida de nuestro software de dispositivo médico acorde a los estándares actuales, sin inhibir el avance.
UNE-EN ISO 14971: Aplicación de la gestión del riesgo a los dispositivos médicos
Preparada para realizar la gestión de riesgos de dispositivos médicos, esta norma ayuda a los fabricantes y desarrolladores de software a identificar todos los peligros asociados con un dispositivo médico y a poder controlar esos riesgos.
ISO 80002-2: Validación software para sistemas de calidad de productos sanitarios
Norma dirigida para la validación de software utilizado en sistemas de calidad de productos sanitarios en conformidad a lo establecido por la EN ISO 13485:2016. Aplica por ello al software utilizado en la gestión de calidad, producción y prestación del servicio, así como en instrumentos de medida.
Controles del ciclo de vida. ISO/TR 80002-2:2017
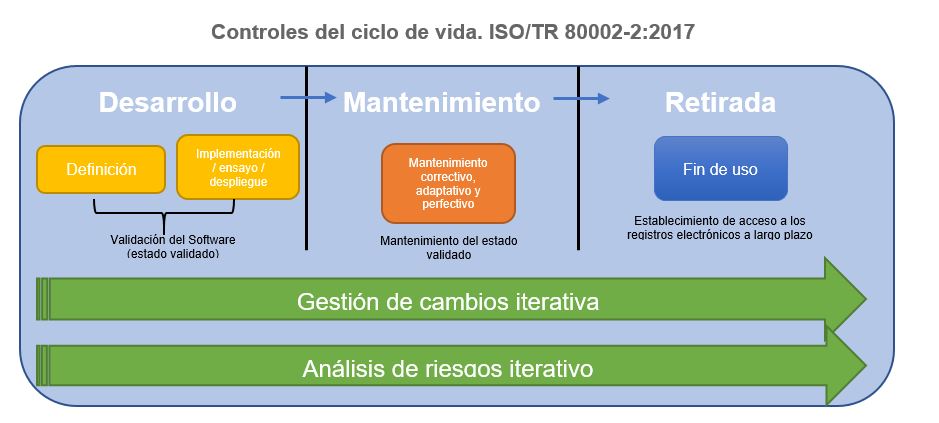
Normativas de referencia de productos sanitarios
Las empresas del sector sanitario deben disponer de un sistema de gestión de calidad que asegure la validación de los sistemas informáticos según las normativas de referencia de aplicación:
- Medical Device Regulation (MDR): Reglamento (UE) 2017/745 sobre los productos sanitarios.
- In Vitro Diagnostic Regulation (IVDR): Reglamento (UE) 2017/746 de Diagnóstico In Vitro.
- FDA Code of federal regulations / Food and Drug Administration: 21 CFR Part 820 – Quality Management Systems (Quality System regulation (QSR))
- Normas ISO:
- UNE-EN-ISO 13485 Productos sanitarios. Sistemas de gestión de la Calidad. Requisitos para fines reglamentarios (ISO 13485:2016)
- ISO/TR 80002-2:2017 Medical device software – Part 2: Validation of software for medical device quality systems
- UNE-EN 62304:2007/A1:2016 Software de dispositivos médicos. Procesos del ciclo de vida del software.
- UNE-EN ISO 14971:2019 Dispositivos médicos/productos sanitarios. Aplicación de la gestión del riesgo a los MD.
- UNE-EN 82304-1:2017 Software sanitario. Parte 1: Requisitos generales para la seguridad de los productos (Ratificada por la Asociación Española de Normalización en octubre de 2017.)
- UNE-CEN ISO/TS 82304-2:2021 Software sanitario. Parte 2: Apps de salud y bienestar. Calidad y confiabilidad (ISO/TS 82304-2:2021) (Ratificada por la Asociación Española de Normalización en septiembre de 2021).
- UNE-EN ISO 14155:2021 Investigación clínica de productos sanitarios para humanos. Buenas prácticas clínicas. (ISO 14155:2020).
- ICH E6 (R2) sobre Buenas Prácticas Clínicas (estándar internacional adoptado por la EMA para el diseño, realización, registro y reporte de ensayos clínicos que involucren la participación de sujetos humanos).
- Normas sobre validación de sistemas informáticos (FDA, ISPE) y Data Integrity:
- 21 CFR Part 11: Criterios dirigidos por FDA para asegurar que los registros y firmas electrónicas son fiables y equivalentes a los registros y firmas escritas en papel.
- Guía GAMP5: Guía desarrollada por la ISPE que promueve un ciclo de vida del SI basado en buenas prácticas. Clarifica las responsabilidades y roles entre la industria farmacéutica y los proveedores de sistemas informáticos.
- Guía GAMP sobre IT Infrastructure Control and Compliance: Guía desarrollada por la ISPE que incluye la orientación para el control adecuado de la infraestructura informática (nuevas tecnologías de virtualización, sistemas/plataformas cloud / XaaS, seguridad, servidores, redes).
- Data Integrity: Guías específicas de la FDA, MHRA, WHO, PIC/S, EMA, GAMP para asegurar la veracidad, consistencia, trazabilidad y disponibilidad de los datos durante todo el ciclo de vida de los datos.
¿Qué ofrece Trescal?
Desde TRESCAL ofrecemos servicios de soporte relativos al ciclo de vida del sistema informático (sistema de gestión de calidad/producción), y/o del software de dispositivo médico según las normativas de aplicación, ofreciendo consultoría específica en las siguientes materias:
- Establecimiento de los requerimientos técnicos y reguladores.
- Revisión del diseño.
- Proceso de desarrollo y mantenimiento del software de dispositivo médico
- Evaluaciones de Sistemas informáticos y Data Integrity (21 CFR Part 11, DIRA, data mapping, GAP analysis, data integrity remediation plan)
- Desarrollo del VMP / VP.
- Establecimiento de estrategias.
- Desarrollo de los expedientes de validación
- Cualificacion de Infraestructura IT (QP, RA, DQ, IQ, OQ, PQ, gestión de cambios y revisión periódica)
- Evaluación de proveedores de SW / tecnológicos.
- Acompañamiento a proveedores en pruebas de aceptación.
- Preparación de procedimientos de gestión y mantenimiento del sistema informático.
- Actividades de formación dirigida.
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría en sistemas de calidad de productos sanitarios, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de equipos, calibraciones, hasta servicios de soporte regulador y registro. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del proyecto de validación, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
 Trescal Derby CMM Machine
Trescal Derby CMM Machine
 TECHNICAL_BNL_ANVERS_2021_TORQUE-3-1
TECHNICAL_BNL_ANVERS_2021_TORQUE-3-1
 Solutions
Solutions iStock_000021513420_Large
iStock_000021513420_Large IMG-20220117-WA0007
IMG-20220117-WA0007 acuerdo marquesme2
acuerdo marquesme2 Control Systems Qualification
Control Systems Qualification imagen-1080×595
imagen-1080×595