Madrid – 17 de Octubre, 2022 – Trescal anuncia la adquisición de Tecnoprocesos en España.
Fundada en 1995 en Barcelona, Tecnoprocesos cuenta con un laboratorio en Sant Cugat (cerca de Barcelona), que añade 1.6M€ al total de facturación y cuenta con 15 trabajadores.
Tecnoprocesos es una empresa especializada en la cualificación de salas limpias, cabinas de flujo laminar, bioseguridad y aspiración, así como redes de gases a presión. Esta adquisición permite a Trescal seguir expandiéndose en el sector lifesciences.
Trescal ha llevado a cabo 43 adquisiciones desde que OMERS es accionista mayoritario en abril 2018.
SOBRE TRESCAL
Trescal es especialista en metrología global y ofrece una amplia gama de servicios de calibración, verificación y prueba de equipos. Trescal está presente en 27 países: Alemania, Australia, Austria, Bélgica, Brasil, Canadá, Corea del Sur, Dinamarca, España, Estados Unidos, Francia, Italia, Luxemburgo, Malasia, Marruecos, México, Nueva Zelanda, Países Bajos, Portugal, Singapur, Suecia, Suiza, República Checa, Rumania, Reino Unido y Túnez. Más allá de sus servicios técnicos (verificación, calibración y reparación de instrumentos T&M), Trescal aporta su experiencia en la adquisición e implementación de sistemas de medición y soluciones de gestión de activos metrológicos. La empresa también ofrece formación y asesoramiento técnico. Trescal emplea a 3.900 personas en una red de más de 160 laboratorios de calibración. Trescal tiene una cartera de 70.000 clientes en las industrias de defensa, aeroespacial, aeronáutica, automotriz, energética, electrónica, telecomunicaciones, química y farmacéutica. Su facturación proforma es de 350 millones de euros. www.trescal.com.
Hands placing last piece of a Puzzle
Concepto Pharma 4.0. Evolución y tendencias de los sistemas informáticos, y transformación digital
La industria 4.0. brinda nuevas oportunidades para la industria Life Science, pero dichas tecnologías deben ser afrontadas con un modelo apropiado basado en el pensamiento crítico, en donde cada vez más el proveedor tecnológico juega un papel relevante.
Introducción
La Industria 4.0, que representa la cuarta revolución industrial, ha llegado a todos los ámbitos de la industria, y el sector farmacéutico y afín, no es una excepción. La presión por la innovación y la eficiencia está haciendo que las compañías farmacéuticas reconsideren la forma en que operan en todos sus ámbitos. Con motivo de la rápida evolución en la tecnología existente, varias tendencias están conduciendo a una digitalización y automatización tanto de los procesos productivos, de laboratorio, logística, gestión de calidad, así como los relacionados con el entorno de la investigación, ensayos clínicos y la Farmacovigilancia. Las tecnologías de Industria 4.0 están revolucionando y revolucionarán muchas áreas, y la forma en que gestionamos la calidad de nuestros datos.
Entre las tendencias y tecnologías clave de la Industria 4.0 se encuentran:
Big Data, Análisis predictivo, centros operativos conectados
La Industria 4.0 representa la era de la interconexión inteligente de los procesos (networking) para el mejor tratamiento de los datos y así ofrecer una mejor productividad, gestión de los recursos, optimización de los procesos y toma de decisiones, escalabilidad y flexibilidad en tiempo real de forma personalizada. Esta capacidad en el tratamiento de los datos para la mejora en continuo de nuestros procesos, marca la gran diferencia respecto a la automatización de los procesos que representaba la Industria 3.0 de los sistemas del pasado.
A medida que ha ido avanzando la tecnología, los procesos de negocio han ido cobrando mayor complejidad, con una mayor presencia de herramientas digitales avanzadas que permiten romper comportamientos o usos no adecuados con el foco en la mejora en continuo de nuestros procesos, eliminar errores humanos, reducir costes y retrasos, y garantizar la conformidad reguladora de los procesos, sistemas y por lo tanto de los productos.
En las últimas décadas hemos ido pasando de procesos manuales basados íntegramente en papel, a procesos digitales con registros electrónicos y firmas electrónicas, sistemas de computación en la nube a, por último, sistemas inteligentes con capacidad de análisis predictivos para la toma de decisiones en tiempo real.
Imagen 2
¿Qué se requiere de los sistemas informáticos?
Los sistemas informáticos actuales deben ser conformes al entorno regulador GxP y de Data Integrity, y deben ser escalables, flexibles, así como facilitar nuestro trabajo, optimizando flujos, y recursos, para que el tratamiento de la información reguladora y la toma de decisiones esté basada en información objetiva y veraz, habiendo eliminado del proceso de decisión, la incertidumbre asociada. Para ello, la aplicación de medidas de control a través del uso del conocimiento, así como la implantación de medidas organizativas son puntos relevantes.
Por todo lo anterior, a la hora de elegir nuestro sistema informático, debemos valorar que éste disponga o cumpla con los siguientes requisitos:
Comprensivo, ágil, intuitivo: Interfaz de usuario fácil e intuitiva adaptable a los flujos del proceso. Búsqueda y consulta ágil de la información.
Facilitador de la toma de decisión basada en el riesgo.
Dispuesto con conexión global (datos, procesos y personas).
Visión holística de los procesos y flujos.
Conectado con otros sistemas informáticos en tiempo real: Integridad ágil y efectiva con otros sistemas informáticos (ej.: ERP, LIMS, etc.), así como infraestructura IT (ej.: BBDD, Directorio activo, servidor de correo, Office, y buscadores de información).
Herramientas avanzadas de predicción de datos (ej.: AI, ML).
Comportamiento predictivo.
Escalable, flexible, dinámico, inteligente: Adaptación modular, extensible al entorno. Funciones analíticas y de reporte. Datos actualizados en tiempo real.
Mejora de los flujos de trabajo. Catalizador de mejoras (promotor de la mejora continua)
A su vez, nuestro sistema informático debe estar diseñado, desarrollado y mantenido dentro de un sistema de gestión de calidad (SGC), por lo que es importante incluir los siguientes requisitos:
Experimentado dentro de un SGC: Historia del producto/solución dentro del sector.
Desarrollado e implementado bajo un modelo comprensible (GAMP5).
Validación: Solución Validada. Paquete de validación, y mantenimiento del estado de control.
Seguro: Medidas de seguridad de protección de datos para asegurar su integridad y privacidad. Entorno web seguro (en el caso de sistemas XaaS[1]).
Conforme desde el inicio: Marco regulador GxP, Data Integrity (incluyendo 21 CFR Part 11, Anexo 11 EU GMP).
Es importante reconocer además que la digitalización y las tecnologías emergentes en la fabricación y el control pueden presentar ciertos desafíos. Por ello, cabe destacar la importancia de la aplicación del QRM (Quality Risk Managament) en el establecimiento de sus requisitos y diseño, teniendo en consideración la transferencia de tecnología de los procesos de producción avanzados, los métodos analíticos, así como en los métodos avanzados de análisis de datos durante todo el ciclo de vida de nuestros sistemas informáticos.
Nuevo enfoque de calidad y relevancia del proveedor tecnológico
El marco regulador existente está siendo revisado en la actualidad (ej.: segunda edición de la GAMP5, nuevo enfoque CSA[2] de la FDA, nueva revisión de la ICH Q9 (R1)) para abordar la creciente importancia de los proveedores de servicios, los enfoques en el desarrollo de software y el uso más generalizado de nuevas tecnologías, herramientas de software y automatización. El uso del pensamiento crítico con los conocimientos y la experiencia para definir los enfoques apropiados son puntos relevantes a tener en cuenta.
Las nuevas tecnologías de AI/ML requieren además de modelos para determinar la estrategia y nivel de esfuerzo de las actividades de validación para poder proporcionar una justificación para la distinción entre los niveles de validación en función de la autonomía y diseño de control del modelo de IA.
Guías de la ISPE como, la GAMP® RDI Good Practice Guide: Data Integrity by Design, la GAMP® Good Practice Guide: Enabling Innovation – Critical Thinking, Agile, IT Service Management, o la GAMP® Good Practice Guide: IT Infrastructure Control and Compliance(Second Edition) intentan clarificar y dotar a la industria de escenarios de referencia relativos a las implicaciones de las diversas tecnologías de la industria 4.0 cada vez más comunes, para el correcto ciclo de vida tanto de los sistemas informáticos, infraestructura IT y/o de los datos GxP.
El nuevo enfoque de calidad pretende aclarar la interpretación de los requisitos regulatorios con el entorno tecnológico actual, para cambiar el foco de la documentación al pensamiento crítico durante todo el ciclo de vida del sistema informático y de su infraestructura de IT que lo soporta.
El objetivo, con este nuevo marco normativo, es ayudar a las empresas reguladas a facilitar el mejor uso de los recursos y fomentar la aplicación de prácticas adecuadas y proporcionadas a QRM, la calidad del producto, la seguridad del paciente y la integridad de los datos.
Imagen 3
El papel del proveedor tecnológico
Cuánto más complejo sea nuestro sistema informático, y mayor impacto posea en nuestras funciones de negocio, mayor relevancia cobra nuestro proveedor para el éxito de nuestro proyecto de IT. Por ello, es importante tener en consideración los siguientes puntos en la inter-relación con nuestro proveedor de IT y su papel como experto en la materia (SME):
Aprovechamiento del proveedor como experto para el incremento del cumplimiento con las perspectivas reguladoras mediante definición de un modelo de ciclo de vida experimentado: Pensamiento crítico para determinar tecnología, metodología y estrategia (GAMP5, Agile, IT Service Management).
Seguimiento de estándares y marco regulador GxP, Data Integrity, ISO, Buenas Prácticas Documentales, GEP (Good Engineering Practices), Sistema de gestión de calidad IT, seguridad de la información, ITIL (Information Technology Infrastructure Library).
Acompañamiento del proveedor en la definición de requisitos facilitando mejoras, automatización de procesos y optimización de tareas para mayor agilidad y eficiencia. Actividades coordinadas iniciadas tempranamente (espíritu de equipo cliente/proveedor).
Cooperación interfuncional y efectiva entre el lado técnico y calidad.
Optimización de tiempos y recursos para el logro de la conformidad: Reducción de costes y tiempos necesarios para conseguir un sistema conforme. Evitar errores.
Resolución de dudas y guía durante todo el proceso que implicará en el cliente en un mayor control del proceso.
Generación de las especificaciones del sistema: Especificaciones de configuración (CS, Configuration Specification), Especificaciones Funcionales (FS, Functional Specification).
Provisión del paquete de verificación/validación: Informes de instalación y configuración y puesta en servicio, paquete de pruebas de validación según modelo de servicio requerido y acordado (generación de las evidencias documentales de soporte de valor añadido a la calidad de las pruebas para la verificación activa de búsqueda de defectos).
Soporte en la gestión de los cambios, configuraciones, problemas, servicios de IT según responsabilidad del modelo de servicio contratado.
Soporte continuado del proveedor en formaciones.
Por todo lo anterior, la competencia y la fiabilidad del proveedor son factores claves a la hora de seleccionar un producto o proveedor de servicios. Por ello, es importante realizar una evaluación con estándares adecuados del proveedor de SW o de servicios IT, ya sea para suministrar, instalar, configurar, integrar, validar, mantener, modificar o conservar un sistema informático o un servicio de IT relacionado. Por ello, debemos contar con procedimientos específicos de evaluación de proveedores tecnológicos que nos permitan conocer si dicho proveedor será o no capaz de cumplir con nuestros requisitos de negocio, reguladores y de integridad de datos. Dichos proveedores deben garantizar la validación continua del software y/o de cualificación de la infraestructura IT relativa, según corresponda, según los acuerdos establecidos[3].
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de infraestructura IT. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución de su proyecto de digitalización desde el punto de vista de calidad, y asegurar el ciclo de vida de su sistema informático y/o infraestructura IT, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros.
[1] XaaS: “X” as a Service. “X” como un Servicio, donde “X” puede ser: “Platform” PaaS (Platform as a Service); IaaS (Infrastructure as a Service); SaaS (Software as a Service).
[2] Guía borrador “CSA Computer Software Assurance for Production and Quality System Software” del centro CDRH (Center for Devices and Radiological Health).
[3] Los departamentos de informática (IT) deben considerarse análogamente a los proveedores de IT, en caso que nuestro sistema informático sea desarrollado y/o mantenido internamente.
Hands placing last piece of a Puzzle
La FDA publica su borrador guía sobre CSA “Computer Software Assurance” para revisión a comentarios
En los últimos años, los avances tecnológicos en los procesos de fabricación, incluyendo la automatización, la digitalización, la robótica, así como los entornos y metodologías de test, han permitido a los fabricantes reducir fuentes de error, optimizar recursos, y reducir el riesgo en la salud del paciente. La FDA reconoce el potencial de estas tecnologías para brindar beneficios significativos para mejorar la calidad, la disponibilidad y la seguridad de los dispositivos médicos, y ha emprendido varios esfuerzos para ayudar a fomentar la adopción y el uso de dichas tecnologías. Dentro de este marco, se publica el nuevo borrador guía como referente para el sector Life Science sobre el nuevo enfoque ”Computer Sofware Assurance”.
Nuevo enfoque para la garantía de calidad del software
El nuevo borrador guía sobre Validación del software “Computer Software Assurance for Production and Quality System Software” recomienda que la «garantía de la calidad del software» se centre en prevenir la introducción de defectos en el proceso de desarrollo de software y fomenta el uso de un enfoque basado en el riesgo para establecer la confianza conforme el software es adecuado para su uso previsto.
Con esta guía la FDA pretende proporcionar recomendaciones sobre la garantía del software informático involucrado en los procesos de producción o del sistema de calidad de un dispositivo médico. Esta guía tiene por objeto:
Describir la «garantía del software informático» como un enfoque basado en el riesgo para establecer la confianza en la automatización utilizada para los sistemas de producción o calidad, e identificar dónde puede ser apropiado un rigor adicional.
Describir diferentes métodos y actividades de test aplicables para establecer la garantía del software informático y proporcionar así evidencia objetiva para cumplir con los requisitos reguladores.
Periodo de consulta
Se establece un periodo de consulta para el envío de comentarios y sugerencias de 60 días a la publicación en el Registro Federal del aviso de disponibilidad de la guía preliminar, por lo que se pueden enviar comentarios y sugerencias hasta el 13 de noviembre 2022.
Aproximación basada en el riesgo
En el borrador guía, la FDA describe un marco de riesgo de aseguramiento del software que incluye la evaluación del uso previsto del sistema informático basado en el riesgo para identificar las actividades de aseguramiento correctas basadas en su nivel de riesgo y crear un registro para documentar que el sistema funcione según lo previsto.
La FDA hace una distinción en la guía entre el software que es parte directa del proceso de producción y el software que respalda el sistema de calidad. Se reconoce además que el software puede tener más de un uso previsto, lo que puede conllevar diferentes riesgos según sus características, funciones y operaciones dentro del proceso de producción o del sistema de calidad.
“FDA recommends that manufacturers document their decision-making process for determining whether a software feature, function, or operation is intended for use as part of production or the quality system in their Standard Operating Procedures (SOPs)”.
Una vez el software ha sido clasificado debe emplearse un análisis basado en el riesgo para identificar las actividades de aseguramiento apropiadas para el software. Todo ello, incluye la identificación de peligros que pudieran impactar en el funcionamiento previsto del sistema y por lo tanto en la producción o el sistema de calidad (riesgo de proceso) o causar un daño en el paciente (seguridad).
“A feature, function, or operation that could lead to severe harm to a patient or user would generally be high device risk. In contrast, a feature, function, or operation that would not foreseeably lead to severe harm would likely not be high device risk.”
Entre las actividades de aseguramiento a aplicar, las pruebas “Scripted Testing” con instrucciones previas escritas de casos de test (como son los test tradicionales de una validación) pueden ser más apropiadas para los sistemas de mayor riesgo, al ser este tipo de pruebas de mayor rigor, mientras que las pruebas “Unscripted Testing” sin instrucciones previas escritas de casos de test y de menor carga documental (tales como pruebas ad-hoc, adivinación de errores, pruebas exploratorias o una combinación de las mismas) pueden ser más apropiadas para sistemas de menor riesgo (pruebas menos planeadas, y limitadas). En este sentido, las pruebas “Unscripted Testing” pueden ser las precursoras de las “Scripted Testing” más formales y rigurosas de estadios de prueba más maduros y formales dentro del proceso de desarrollo y verificación del software.
Los fabricantes también deben asegurarse de crear un registro apropiado para documentar que la característica, función u operación del software funciona según lo previsto. El registro apropiado debe incluir el uso previsto, la determinación de los riesgos y la documentación de las actividades de aseguramiento a realizar. Sobre este asunto, la nueva guía pretende crear una nueva oportunidad para racionalizar la documentación, al aplicar el pensamiento crítico y la gestión de riesgos con relación al impacto del sistema en la seguridad del paciente y el producto dentro de un enfoque de garantía de calidad.
“Documentation of assurance activities need not include more evidence than necessary to show that the software feature, function, or operation performs as intended for the risk identified. FDA recommends the record retain sufficient details of the assurance activity to serve as a baseline for improvements or as a reference point if issues occur”.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, validación de sistemas, cualificación de equipos y calibración. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten:
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Proceso de desarrollo y mantenimiento del software de dispositivo médico.
Evaluaciones de Sistemas informáticos y Data Integrity: verificación 21 CFR Part 11, Data Integrity Risk Assessment (DIRA), data process mapping, data integrity remediation plan, orphan data check).
Desarrollo del VMP / VP.
Revisión y evaluación de metodologías y actividades de test. GAP análisis, AMFE.
Desarrollo de procedimientos normalizados de trabajo.
Desarrollo de los expedientes de validación.
Pruebas de aceptación FAT / SAT / Comisionado.
Cualificacion de Infraestructura IT (QP, RA, DQ, IQ, OQ, PQ, gestión de cambios y revisión periódica).
Evaluación de proveedores de SW / tecnológicos.
Acompañamiento a proveedores en pruebas de aceptación.
Preparación de procedimientos de gestión y mantenimiento del sistema informático.
Implantación de Sistemas de Calidad IT. Sistemas de Calidad para desarrolladores de software.
Reevaluaciones periódicas.
Procesos de migración de datos.
Evaluación ante la retirada de un sistema informático.
Plan de Transformación CSA.
Actividades de formación dirigida.
Si está evaluando la necesidad de contar con un proveedor experto que le acompañe en este proceso de transformación al nuevo enfoque CSA desde la órbita de calidad, y /o asegurar y agilizar la correcta consecución del ciclo de vida de su sistema informático y/o infraestructura IT, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
iStock_000020268060Large
Nuevo anexo 1 EU GMP y estrategia de control de la contaminación (CCS). ¿Estás preparado?
El entorno regulador y tecnológico en la fabricación de medicamentos estériles avanza, y por ello, es importante estar preparado y contar con proveedores expertos que acompañen a los fabricantes en este proceso de transformación. El control de la contaminación y la elaboración de una estrategia de control de la contaminación es un concepto relevante que define la nueva versión del Anexo 1.
Estrategia de control de la contaminación
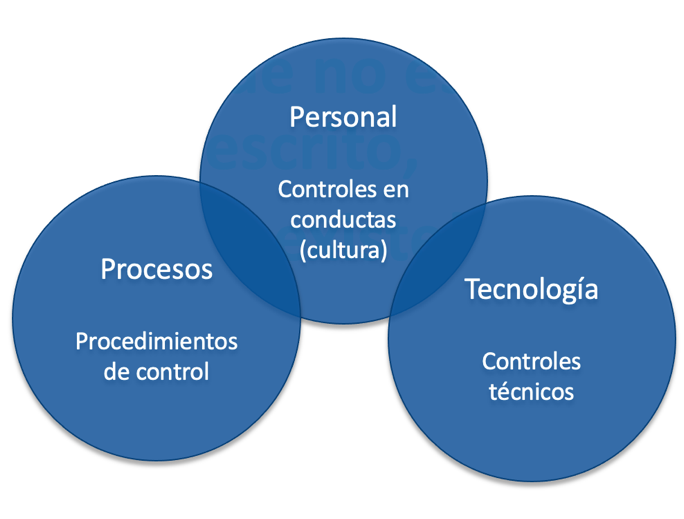
El Anexo 1 GMP de la EU sobre «Fabricación de medicamentos estériles« ha sido revisado a finales de agosto tras varios años de ansiada espera. Tal y como mencionábamos en una anterior noticia, la nueva versión hace foco en una mejor aplicación del proceso de gestión de los riesgos (ICH Q9 – Quality Risk Management) y del sistema de calidad farmacéutico (ICH Q10 – Pharmaceutical Quality System) para asegurar un mayor control de la contaminación. En este ámbito, el nuevo anexo enfatiza la implementación de una estrategia de control de la contaminación (CCS, Contamination Control Strategy, por sus siglas en inglés) para definir los puntos críticos de control y evaluar la eficacia de todos los controles (de diseño, procedimiento, técnicos y organizativos), así como las medidas de seguimiento empleadas.
“Contamination Control Strategy (CCS) – A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control.”(Glossary)
El nivel de esfuerzo y de detalle de la estrategia de control de la contaminación debe ser proporcional al tipo de proceso y producto. Debe considerarse que el nuevo Anexo 1 no solo provee directriz sobre la fabricación de productos «estériles» sino que también brinda orientación sobre la fabricación de los productos “no estériles”:
“The intent of the Annex is to provide guidance for the manufacture of sterile products. However, some of the principles and guidance, such as contamination control strategy, design of premises, cleanroom classification, qualification, validation, monitoring and personnel gowning, may be used to support the manufacture of other products that are not intended to be sterile such as certain liquids, creams, ointments and low bioburden biological intermediates, but where the control and reduction of microbial, particulate and endotoxin/pyrogen contamination is considered important. Where a manufacturer elects to apply guidance herein to non-sterile products, the manufacturer should clearly document which principles have been applied and acknowledge that compliance with those principles should be demonstrated.” (1 Scope)
Por otro lado, la estrategia de control de la contaminación debe revisarse activamente y, ser actualizada correspondientemente para impulsar la mejora continua de los métodos de fabricación y control. Su eficacia debe formar parte de la gestión de los procesos de revisión periódica.
Elementos a considerar dentro de la estrategia de control de contaminación
Tal y como menciona el Anexo 1, los elementos a considerar dentro de un CCS deben ser (pero no limitados a éstos) los siguientes:
Diseño de la planta y de los procesos, incluyendo la documentación asociada.
Locales y equipos.
Personal.
Servicios.
Controles de materias primas, incluyendo los controles en proceso.
Envases de productos y cierres.
Aprobación de proveedores.
Gestión de actividades subcontratadas y disponibilidad/transferencia de la información crítica entre las partes (ej.: servicios de esterilización).
Proceso de gestión de riesgos.
Validación del proceso.
Validación de procesos de esterilización.
Mantenimiento preventivo: mantenimiento de equipos, servicios e instalaciones (mantenimiento planificado y no planificado) para asegurar que no haya riesgo adicional de contaminación.
Limpieza y desinfección.
Sistemas de monitorización, incluyendo una evaluación de la viabilidad de la introducción de métodos alternativos científicamente sólidos que optimicen la detección de la contaminación ambiental.
Mecanismos de prevención: análisis de tendencias, investigación detallada, determinación de la causa raíz, acciones correctivas y preventivas (CAPA) y la necesidad de herramientas de investigación exhaustivas.
Mejora continua en base a la información derivada de lo anterior.
Desarrollo, documentación y mantenimiento de la estrategia de control de la contaminación
Los fabricantes de productos farmacéuticos, así como sus proveedores son conscientes que la contaminación puede provocar pérdidas de producto, o pueden representar un riesgo importante sobre la protección de la salud pública.
Es por ello crucial, la preparación de un documento CCS que brinde una descripción general de la totalidad de las medidas de control de la contaminación y su vínculo con la estrategia general CSS. El proceso de implantación de la estrategia de control de la contaminación se puede resumir en las siguientes etapas:
Etapa 1: Desarrollo (o revisión/mejora) de la estrategia de control de la contaminación (ej.: revisión de requisitos y necesidades, desarrollo de un Gap análisis para la detección de vulnerabilidades y su corrección).
Estado 2: Compilación de los documentos CCS (documentar todas las medidas, incluyendo procedimientos, QRM, controles, reportes de cualificación y validación, etc.) para el desarrollo del documento CSS.
Etapa 3: Evaluación y mantenimiento del CCS (revisiones en continuo, revisiones periódicas).
Tal y como especifica el propio Anexo 1, el desarrollo del CCS requiere de conocimiento técnico y de proceso detallado. El fabricante debe poseer suficiente conocimiento y experiencia sobre los productos fabricados, los equipos y los métodos de fabricación e ingeniería empleados para evaluar su impacto en la calidad del producto.
“The development of the CCS requires detailed technical and process knowledge. Potential sources of contamination are attributable to microbial and cellular debris (e.g. pyrogen, endotoxin) as well as particulate (e.g. glass and other visible and sub-visible particles).” (2 Principle)
Cobra relevancia el soporte de los proveedores y su correcta evaluación previa como expertos en la materia para el correcto acompañamiento durante todo el ciclo de vida de los sistemas y equipos clave. Ej.: proveedores de equipos y componentes críticos, esterilización, desinfectantes y detergentes, bioindicadores, ingenierías de servicios farmacéuticos (agua, vapor, gases), mantenimiento, consultoría y cualificación.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, validación de sistemas, cualificación de equipos y calibración. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten:
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Gap análisis de la estrategia actual de control de la contaminación.
Evaluación de proveedores.
Cualificación de salas limpias.
Cualificación de servicios farmacéuticos.
Cualificación de equipos de esterilización.
Establecimiento y mantenimiento de los planes de verificación de salas limpias.
Desarrollo del plan de estrategia de control de la contaminación.
Validaciones de proceso.
Validaciones de limpieza.
Desarrollo de los expedientes de validación / cualificación.
Acompañamiento a proveedores en pruebas de aceptación.
Revisiones periódicas.
Calibración instrumental.
Actividades de formación dirigida.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta implantación de su estrategia de control de contaminación, así como la correcta validación/cualificación y estado de control de sus instalaciones/equipos desde la órbita de calidad, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
GMP Qualification and validation
PUBLICAÇÃO DO ANEXO 1 EU GMP: ACTUALIZAÇÃO DO NOVO AMBIENTE REGULAMENTAR E DE FABRICO
Introdução e base para a nova revisão
A fim de atualizar o atual ambiente regulamentar e tecnológico no fabrico de medicamentos esterilizados, o Anexo 1 foi revisto. Após quase 5 anos de espera desde o primeiro projeto em 2017 e mais 2 anos desde o segundo projeto em 2020 com a revisão dos respetivos comentários públicos, a Comissão da UE publicou em 22 de Agosto o tão esperado Anexo 1 das GMP da UE sobre «Fabricação de medicamentos esterilizados».
O novo anexo esclarece como as empresas fabricantes podem tirar partido das novas possibilidades resultantes da implementação de uma melhor compreensão do processo de gestão de riscos (ICH Q9 – Gestão de Riscos de Qualidade) e do sistema de qualidade farmacêutica (ICH Q10 – Sistema de Qualidade Farmacêutica). Inclui também novos desenvolvimentos tecnológicos que exigiram a revisão do Anexo 1. Por exemplo, nova secção sobre Estratégia de Controlo de Contaminação (CCS), e novas secções para acomodar os recentes avanços na tecnologia de fabrico estéril, tais como Sistemas de Barreiras de Acesso Restrito (RABS) e isoladores.
A revisão do Anexo 1 também tem em conta as alterações noutros capítulos e anexos de GMP e/ou liga-os para controlo posterior (por exemplo, Capítulo 1 sobre Sistema de Qualidade Farmacêutico, Anexo 4 sobre Fabrico de medicamentos veterinários que não medicamentos veterinários imunológicos, Anexo 12 sobre Utilização de Radiações Ionizantes no fabrico de medicamentos, Anexo 15 sobre Qualificação e Validação, Anexo 17 sobre Libertação Paramétrica), bem como outros regulamentos relacionados (ISO 14644).
Principais alterações em relação ao projeto para 2020
Em comparação com o projeto anterior de 2020, a estrutura básica do Anexo 1 permaneceu inalterada, mas a nova revisão é mais abrangente, aumentando de 52 para 58 páginas, e há numerosas supressões, resumos e novas inserções em muitos capítulos.
Por exemplo, o subcapítulo «Tecnologias de Barreiras» no Capítulo 4 «Instalações» é quase duplicado para tratar em maior detalhe a utilização de luvas e materiais, bem como métodos de descontaminação em RABS e isoladores separadamente, e os subcapítulos «Form-Fill-SEAL (FFS)» e «Blow-Seleal» (equipamento utilizado no fabrico de produtos terminados esterilizados) no Capítulo 8 «Produção e Tecnologias Específicas» quase triplicam em conteúdo.
Prazos
O novo Anexo 1 entrará em vigor a 25 de Agosto de 2023, ou seja, um ano após a publicação no Eudralex Volume 4, com exceção do ponto 8.123 (esterilização em liofilizadores), que será aplicável a 25 de Agosto de 2024.
Trescal
TRESCAL é um prestador de serviços global para a indústria das Ciências da Vida. Oferece serviços de implementação/consultoria de qualidade, validação de sistemas, qualificação de equipamentos e calibração. O nosso conhecimento específico e transversal permite-nos oferecer um serviço de qualidade global como especialistas na área, onde quer que necessite.
Se estiver a avaliar a necessidade de um fornecedor especializado para assegurar e acelerar a correta realização e validação do seu processo de esterilização, ou para resolver com sucesso o seu projeto de qualificação de sala limpa e/ou equipamento de esterilização a partir da órbita da qualidade, poupando eventos e recursos imprevistos, não perca mais tempo e contacte-nos, teremos todo o prazer em ouvi-lo e juntar-se ao seu projeto.
GMP Qualification and validation
Publicación del anexo 1 EU GMP: Actualización al nuevo entorno regulador y de fabricación
Introducción y bases de la nueva revisión
Con motivo de actualizar el entorno actual regulador y tecnológico en la fabricación de medicamentos estériles, el Anexo 1 ha sido revisado. Tras casi 5 años de espera desde el primer borrador en el año 2017 y más 2 años desde el segundo borrador en el año 2020 con la revisión de sus respectivos comentarios públicos, la Comisión de la EU ha publicado el pasado 22 de agosto, el esperado Anexo 1 GMP de la EU sobre «Fabricación de medicamentos estériles«.
El nuevo anexo clarifica cómo las empresas fabricantes pueden aprovechar las nuevas posibilidades derivadas de la aplicación de una comprensión mejorada del proceso de gestión de los riesgos (ICH Q9 – Quality Risk Management) y del sistema de calidad farmacéutico (ICH Q10 – Pharmaceutical Quality System). También incluye nuevos desarrollos tecnológicos que han hecho necesaria la revisión del Anexo 1. Ej.: nueva sección sobre estrategia de control de la contaminación (CCS, Contamination Control Strategy, por sus siglas en inglés), y nuevas secciones para adoptar los recientes avances en la tecnología de fabricación estéril como son los sistemas de barrera con acceso restringido (RABS, Restricted Access Barrier System, por sus siglas en inglés) y los aisladores.
La revisión del Anexo 1 tiene en cuenta, además, los cambios en otros capítulos y anexos GMP y/o los relaciona para mayor control (ej.: Capítulo 1 sobre Sistema de Calidad Farmacéutico, anexo 4 sobre Fabricación de medicamentos veterinarios distintos de los medicamentos veterinarios inmunológicos, anexo 12 sobre Uso de las radiaciones ionizantes en la fabricación de medicamentos, anexo 15 sobre Cualificación y Validación, anexo 17 sobre Liberación Paramétrica), así como otras regulaciones relacionadas (ISO 14644).
Principales cambios respecto el borrador del año 2020
Comparativamente con el anterior borrador del año 2020, la estructura básica del Anexo 1 se ha mantenido sin cambios, pero la nueva revisión es más completa, pasando de 52 a 58 páginas, y existen numerosas deleciones, resúmenes y nuevas inserciones en numerosos capítulos.
Por ejemplo, el subcapítulo «Barrier Technologies» en el capítulo 4 «Premises» casi se duplica para tratar con mayor detalle el uso de guantes y materiales, así como de métodos de descontaminación en RABS y aisladores por separado, y los subcapítulos «Form-Fill-SEAL (FFS)» y «Blow-Seleal» (equipos utilizados en la fabricación de productos con esterilización terminal) en el capítulo 8 «Production and Specific Technologies» casi triplican su contenido.
Plazos
El nuevo Anexo 1 entrará en vigor el 25 de agosto de 2023, es decir, un año después de la publicación en Eudralex Volumen 4, a excepción del punto 8.123 (esterilización en liofilizadores), que será de aplicación el 25 de agosto de 2024.
Trescal
TRESCALes un proveedor global de servicios para la industria Life Science. Ofrece servicios de implantación/consultoría de calidad, validación de sistemas, cualificación de equipos y calibración. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución y validación de su proceso de esterilización, o afrontar con éxito su proyecto de cualificación de sala limpia y/o equipo de esterilización desde la órbita de calidad, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Hands placing last piece of a Puzzle
Segunda Edición Guía ISPE GAMP® 5: Actualización a los nuevos modelos, requisitos y usos tecnológicos
Introducción y bases de la segunda edición
Tras 14 años de la primera edición de la Guía ISPE GAMP® 5 A Risk-Based Approach to Compliant GxP Computerized Systems, este mes de julio se ha publicado finalmente su segunda y esperada edición.
Las guías GAMP pretenden ofrecer un marco de referencia en buenas prácticas dentro del sector Life Science para garantizar que los sistemas informáticos sean efectivos, confiables y de alta calidad. De esta forma son adecuados para su uso previsto y cumplen con las regulaciones aplicables.
Manteniendo los principios y el marco de la primera edición, la nueva guía ISPE GAMP® 5 actualiza su aplicación habiendo revisado las prácticas, regulaciones y desarrollos tecnológicos actuales, para incluir la mayor relevancia de los proveedores IT de servicios, los enfoques en evolución del desarrollo de software, y el uso ampliado de herramientas de software y automatización. Destaca el uso del pensamiento crítico de los expertos en la materia (SME, subject matter expert por sus siglas en inglés) para poder definir los enfoques más apropiados en las circunstancias específicas en base a su conocimiento y experiencia.
Por lo tanto, la nueva guía ISPE GAMP® 5 incluye y fomenta lo siguiente:
Mayor relevancia de los proveedores de servicios de IT. Incluyendo los proveedores de servicios en la nube (cloud), para alentar a las empresas reguladas a maximizar la participación de los proveedores y así aprovechar su conocimiento, experiencia y documentación, cuando sea posible.
Enfoques en evolución del desarrollo de software, enfatizando que el enfoque de especificación y verificación GAMP no es inherentemente lineal, sino que también admite perfectamente métodos iterativos e incrementales.
Mayor uso de herramientas de software y automatización para lograr un mayor control, calidad y menores riesgos durante todo el ciclo de vida del sistema informático.
Además, la nueva guía, incluye el nuevo enfoque Computer Software Assurance (CSA, por sus siglas en inglés) de la FDA, y clarifica materias referenciando otras guías GAMP RDI sobre integridad de datos (DI, Data Integrity, por sus siglas en inglés). Además, incluye nuevas consideraciones sobre el uso de software open-source (OOS, Open Source Software, por sus siglas en inglés).
Mientras que los enfoques de especificación y verificación, así como el marco general y conceptos clave del ciclo de vida del sistema, y el enfoque de gestión de riesgos de calidad (alineado con la ICH Q9) permanecen sin cambios, el contenido técnico de la guía ha sido revisado. Por ello, los apéndices de la nueva guía han sido actualizados para incorporar las prácticas, requisitos regulatorios y tecnologías actuales, dentro de la evolución tecnológica de la industria del sector Life Science y el marco Pharma 4.0TM.
Por ello, son de nueva creación los siguientes apéndices: D8 (Agile). D9 (Software Tools). D10 (Blockchain). D11 (Artificial Intelligence and Machine Learning – AI/ML, por sus siglas en inglés-). M11 (IT Infrastructure) y M12 (Critical Thinking).
Además, han sido significativamente revisados los siguientes apéndices: D1 (Specifying Requirements), y S2 (Electronic Production Records).
Por el contrario, han sido eliminados los siguientes apéndices: D2 (Functional Specifications) que ha sido combinado junto el apéndice D1 para estar en un único apéndice, O7 (Repair Activity) que ha sido revisado para estar definido en el apéndice O6 (Operational Change and Configuration Management) y S5 (Managing Quality within an Outsourced IS/IT Enviromnment) cuyo contenido ha sido revisado para ser incluido dentro del nuevo apéndice M11 (IT Infrastructure).
A su vez, la nueva guía considera que son áreas sinérgicas, tanto la Gestión del conocimiento, así como el Advancing Pharmaceutical Quality (APQ, por sus siglas en inglés) y la transformación digital Pharma 4.0 TM, siendo la madurez digital y la integridad de datos elementos facilitadores para el establecimiento de una efectiva estrategia de transformación digital.
¿A quién está dirigida?
La nueva guía está destinada tanto a empresas reguladas, como proveedores y reguladores. Es importante aclarar que, por proveedores, se incluyen a proveedores de software, hardware, equipos, servicios de integración de sistemas, servicios IT, tanto internos como externos a la empresa regulada.
Trescal
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de infraestructura IT. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del ciclo de vida de su sistema informático, infraestructura IT, o afrontar con éxito su proyecto de transformación digital desde la órbita de calidad, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
iStock_000016752326Large
UNE-EN ISO 13485:2018
Desde Abril 2022 Trescal dispone de la certificación en ISO13485:2016.
Con el siguiente alcance:
Calibración
Inspección
Mantenimiento
Reparación de equipos electromédicos
Como decisión estratégica se ha optado por adquirir la certificación bajo la norma ISO 13485:2016 (UNE-EN ISO 13485:2018). Productos sanitarios. Sistemas de gestión de la calidad. Requisitos reglamentarios. Eligiendo a ASSI Trescal que sita en Barcelona, como laboratorio poseedor de esta certificación, por su gran actividad en el sector biomédico.
Esta norma establece los requisitos para un sistema de gestión de la calidad de las organizaciones involucradas en las diferentes etapas del ciclo de vida de un producto sanitario. Abarca tanto a fabricantes de estos productos como a proveedores. En el caso de ASSI Trescal, como empresa de servicios de calibración y mantenimiento, se ha elegido voluntariamente cumplir con estos requisitos y certificarnos.
Ha sido muy fácil integrar los requisitos de la norma a nuestro Sistema de Calidad. ASSI Trescal posee un gran conocimiento en el sector al que nos dirigimos y sabe perfectamente cuáles son los requerimientos de sus clientes y los reglamentos aplicables.
Cualificación de la infraestructura IT y estrategia de control en su ciclo de vida.
A medida que las organizaciones han ido dependiendo de los sistemas informáticos para la automatización de sus procesos y actividades reguladas, la infraestructura IT que los soporta ha ido cobrando cada vez mayor relevancia.
Situación actual y cualificación infraestructura IT
En los últimos años ha habido un salto significativo en el ámbito de la Infraestructura de la Tecnología de la Información (IT) que ha dado lugar a una reorientación y actualización de las políticas de validación/cualificación en esta materia. Aunque el anexo 11 de las EU GMP ya hacía alusión a este tema en su principio “La aplicación debe validarse; la infraestructura informatizada (IT) debe cualificarse”, la complejidad tecnológica y los problemas en Data Integrity, han derivado en un mayor aseguramiento del estado de control de la infraestructura IT.
El salto significativo en la Infraestructura IT se resume en los siguientes cambios del modelo tecnológico, muy bien recogidos en la Guía GAMP sobre IT Infrastructure Control and Compliance (agosto 2017):
El uso de tecnologías de virtualización que permiten compartir, combinar y maximizar los recursos.
El uso de sistemas Cloud, incluyendo las aplicaciones GxP «as-a-service» y los modelos de servicios de tipo XaaS: infraestructura como servicio (IaaS), plataforma como servicio (PaaS) y software como servicio (SaaS).
La mayor externalización de actividades IT en proveedores tecnológicos (Outsourcing) y uso de Third party data centers.
Gestión de los riesgos
La gestión de riesgos proporciona un método para identificar aspectos de una manera controlada y sistemática, y debe realizarse para cada sistema y/o proceso de infraestructura relevante. Durante este proceso, es importante el poder dar respuesta a las siguientes preguntas:
¿Qué aplicaciones GxP disponemos?
¿Cuál es la infraestructura IT que soporta estas aplicaciones GxP? ¿Qué plataformas, sistemas y/o componentes IT componen esta infraestructura IT? ¿Cuál es el estado de validación y/o cualificación de dichas plataformas, componentes? ¿Esta cualificación se encuentra correctamente documentada y soportada por un ejercicio de gestión de los riesgos –trazabilidad con los riesgos?
¿Se requieren controles adicionales para el adecuado control de los riesgos existentes incluyendo los riesgos en materia de Data Integrity? ¿Deben implantarte cambios?
Se realizan revisiones periódicas para la evaluación y adecuado control de la infraestructura IT?
Cualificación de la Infraestructura IT
La Cualificación de la Infraestructura IT es la confirmación mediante examen y provisión de evidencias objetivas que las especificaciones de la Infraestructura IT son conformes a los requerimientos del usuario y a su uso previsto, y que todos los requerimientos pueden ser consistentemente cumplidos.
Al tratarse, la Infraestructura IT, en su conjunto de una unidad compuesta por varios procesos, servicios, y plataformas compartidas, es importante abordar la estrategia de cualificación de forma segmentada, o global dependiendo del estudio de gestión de los riesgos previo para evitar retrabajos, y maximizar el esfuerzo de validación/cualificación en aquellas áreas de mayor riesgo.
Sistema de gestión de calidad y mantenimiento del estado cualificado
Es importante disponer de un sistema de gestión de calidad implantado y robusto que asegure el adecuado estado de control de la infraestructura IT. Por ello, es importante el revisar el estado actual de la documentación de nuestra infraestructura IT (Manual de Calidad con la inclusión de los procesos clave de infraestructura IT, Job Descriptions que definan la asignación de roles y responsabilidades, Procedimientos dirigidos al mantenimiento del estado de control y cualificación) por si fuera necesaria completarla y/o actualizarla, y el de rodearnos de personal experto en caso de necesitar ayuda o soporte para su adecuado desarrollo.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de infraestructura IT. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del ciclo de vida de su infraestructura IT, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
iStock_000019934130Large
Sistemas informáticos en la industria Life Science: Consultoría de calidad y Validación
Introducción y marco previo regulador
En los últimos años, la tecnología ha avanzado significativamente, y la industria regulada se ha apoyado en una gran variedad de sistemas informáticos para la realización de sus funciones de negocio. Además, el actual marco normativo se ha ido adaptando para dotar de mejor aseguramiento al estado de control de los sistemas informáticos durante todo su ciclo de vida, con especial foco en la calidad e integridad del dato.
Las guías de Data Integrity de los diferentes autoridades y agencias reguladoras, junto con el nuevo enfoque CSA de la FDA sobre Computer System Assurance, y la consecuente próxima publicación de la segunda edición de la Guía GAMP 5 tras 14 años de su primera edición, hace que el entorno de la industria Life Science ponga de nuevo el foco, aún más si cabe, en la gestión del ciclo de vida de los sistemas informáticos, así como en el ciclo de vida del dato.
Ciclo de vida de los sistemas informáticos
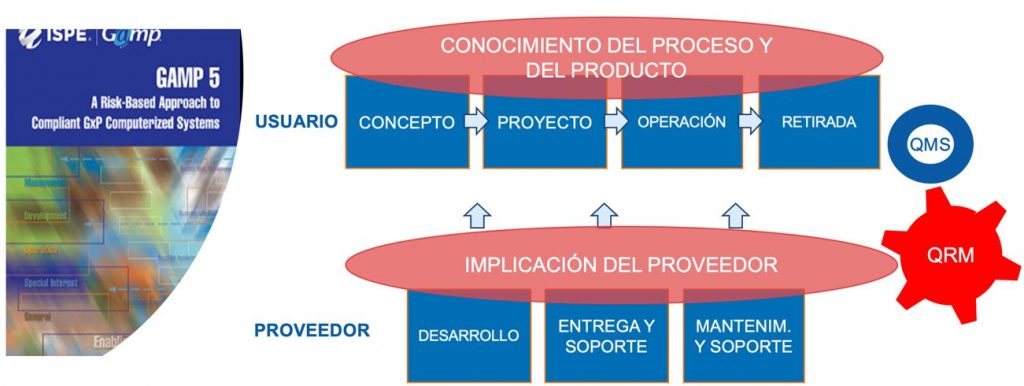
Disponer de un Sistema de Calidad IT es un elemento clave para asegurar el correcto ciclo de vida de nuestro sistema informático. Además. en un entorno cada vez más complejo y de mayor especialización técnica, la implicación del proveedor de la aplicación y/o tecnológico cobra mayor relevancia, por lo que es crucial su adecuada evaluación.
Las fases del ciclo de vida de un sistema informático según la Guía GAMP 5 son las siguientes:
Concepto: Establecimiento de los requerimientos, descripción general y consideraciones de diseño
Proyecto: Planificación, selección del proveedor y de la solución, especificaciones detalladas del sistema, y validación
Operación: Gestión del sistema en su fase operativa (ej.: operación, mantenimiento, gestión de cambios y configuración, revisiones periódicas, incidencias, copia de seguridad, restauración de datos, seguridad lógica y física, plan de contingencia).
Retirada: Retirada, migración, sustitución del sistema
Además, existen procesos o actividades de apoyo que tienen lugar en las fases del ciclo de vida que salen reforzadas inherentemente en el nuevo concepto CSA, como es la gestión de riesgos, la gestión del conocimiento, el pensamiento crítico, las buenas prácticas documentales, y/o la evaluación del proveedor.
Validación de sistemas informáticos
Según la FDA (21 CFR part 11), la validación de un sistema informático es la confirmación mediante examen y provisión de evidencias objetivas que las especificaciones de un sistema informatizado son conformes a los requerimientos del usuario y a su uso previsto, y que todos los requerimientos pueden ser consistentemente cumplidos.
Se trata pues de una actividad destinada a asegurar el uso previsto del sistema informático con el fin de cumplir con el marco regulador aplicable, así como conseguir las autorizaciones y certificaciones necesarias. Esta actividad además nos permite conocer mejor nuestro sistema, sus vulnerabilidades y oportunidades de mejora, no sólo en lo referente a aspectos de calidad, sino también de productividad. Aplicar una buena metodología en nuestro proyecto de validación permite una reducción del coste y tiempo necesarios para conseguir un sistema conforme, garantizando el cumplimiento de los requisitos y de las perspectivas reguladoras desde el inicio.
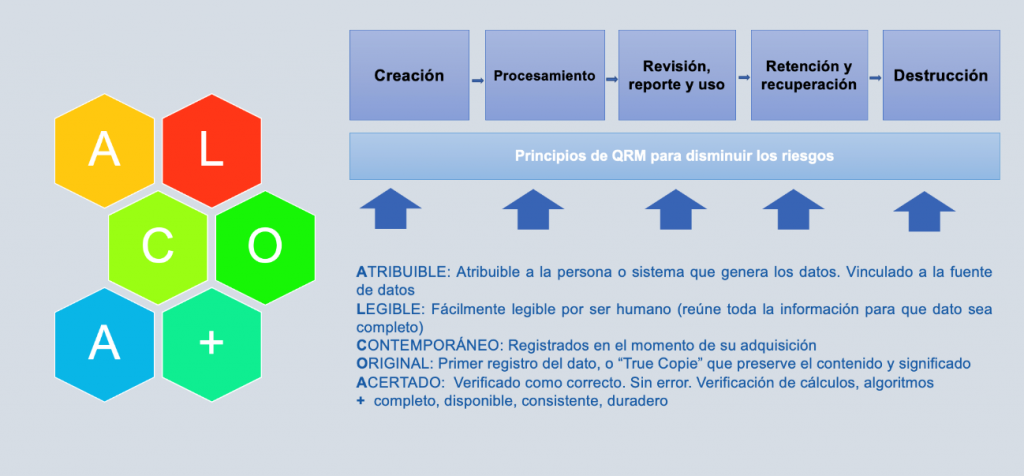
Ciclo de vida del dato
Con el nuevo enfoque Data Integrity, además de asegurar el correcto ciclo de vida del sistema informático, debemos asegurar el ciclo de vida del dato (principio ALCOA+).
Para ello, es crucial el poseer de un sistema maduro que incorpore la Gobernanza de los datos que abarque los siguientes conceptos clave.
Normativas de referencia de sistemas informáticos
Las empresas Life Science deben disponer de un sistema de gestión de calidad que asegure el correcto ciclo de vida de los sistemas informáticos y de sus datos conforme al marco regulador del producto en cuestión:
21 CFR Part 11: Criterios dirigidos por FDA para asegurar que los registros y firmas electrónicas son fiables y equivalentes a los registros y firmas escritas en papel.
Anexo 11 de las EU GMP: Requisitos de la comisión europea para los sistemas informáticos. Cuando un sistema informático reemplace una operación manual, no debe ser en detrimento de la calidad del producto, control del proceso o garantía de calidad. No debe haber un incremento del riesgo total del proceso.
Guía GAMP5: Guía desarrollada por la ISPE que promueve un ciclo de vida del sistema informático basado en buenas prácticas. Clarifica las responsabilidades y roles entre la industria farmacéutica y los proveedores de sistemas informáticos.
Guía GAMP IT Infrastructure Control and Compliance: Guía desarrollada por la ISPE que incluye la orientación para el control adecuado de la infraestructura informática (nuevas tecnologías de virtualización, sistemas/plataformas cloud / XaaS, seguridad, servidores, redes).
Data Integrity: Guías específicas de la FDA, MHRA, WHO, PIC/S, EMA, GAMP para asegurar la veracidad, consistencia, trazabilidad y disponibilidad de los datos durante todo el ciclo de vida de los datos.
Normas específicas de productos sanitarios:
ISO/TR 80002-2:2017 Medical device software – Part 2: Validation of software for medical device quality systems
UNE-EN 62304:2007/A1:2016 Software de dispositivos médicos. Procesos del ciclo de vida del software.
UNE-EN ISO 14971:2019 Dispositivos médicos/productos sanitarios. Aplicación de la gestión del riesgo a los MD.
UNE-EN 82304-1:2017 Software sanitario. Parte 1: Requisitos generales para la seguridad de los productos (Ratificada por la Asociación Española de Normalización en octubre de 2017.)
UNE-CEN ISO/TS 82304-2:2021 Software sanitario. Parte 2: Apps de salud y bienestar. Calidad y confiabilidad (ISO/TS 82304-2:2021) (Ratificada por la Asociación Española de Normalización en septiembre de 2021).
¿Qué ofrece Trescal?
Desde TRESCAL ofrecemos servicios de soporte relativos al ciclo de vida del sistema informático (sistema de gestión de calidad/producción), y/o del software de dispositivo médico según las normativas de aplicación, ofreciendo consultoría específica en las siguientes materias:
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Proceso de desarrollo y mantenimiento del software de dispositivo médico.
Evaluaciones de Sistemas informáticos y Data Integrity: verificación 21 CFR Part 11, Data Integrity Risk Assessment (DIRA), data process mapping, GAP analysis, data integrity remediation plan, orphan data check).
Desarrollo del VMP / VP.
Establecimiento de estrategias.
Desarrollo de los expedientes de validación.
Pruebas de aceptación FAT / SAT / Comisionado.
Cualificacion de Infraestructura IT (QP, RA, DQ, IQ, OQ, PQ, gestión de cambios y revisión periódica).
Evaluación de proveedores de SW / tecnológicos.
Acompañamiento a proveedores en pruebas de aceptación.
Preparación de procedimientos de gestión y mantenimiento del sistema informático.
Implantación de Sistemas de Calidad IT. Sistemas de Calidad para desarrolladores de software.
Reevaluaciones periódicas.
Procesos de migración de datos.
Evaluación ante la retirada de un sistema informático.
Plan de Transformación CSA.
Actividades de formación dirigida.
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de infraestructura IT. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del ciclo de vida de su sistema informático y/o infraestructura IT, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
iStock_000018808720Large
Trescal adquiere Tecnoprocesos en España
Madrid – 17 de Octubre, 2022 – Trescal anuncia la adquisición de Tecnoprocesos en España.
Fundada en 1995 en Barcelona, Tecnoprocesos cuenta con un laboratorio en Sant Cugat (cerca de Barcelona), que añade 1.6M€ al total de facturación y cuenta con 15 trabajadores.
Tecnoprocesos es una empresa especializada en la cualificación de salas limpias, cabinas de flujo laminar, bioseguridad y aspiración, así como redes de gases a presión. Esta adquisición permite a Trescal seguir expandiéndose en el sector lifesciences.
Trescal ha llevado a cabo 43 adquisiciones desde que OMERS es accionista mayoritario en abril 2018.
SOBRE TRESCAL
Trescal es especialista en metrología global y ofrece una amplia gama de servicios de calibración, verificación y prueba de equipos. Trescal está presente en 27 países: Alemania, Australia, Austria, Bélgica, Brasil, Canadá, Corea del Sur, Dinamarca, España, Estados Unidos, Francia, Italia, Luxemburgo, Malasia, Marruecos, México, Nueva Zelanda, Países Bajos, Portugal, Singapur, Suecia, Suiza, República Checa, Rumania, Reino Unido y Túnez. Más allá de sus servicios técnicos (verificación, calibración y reparación de instrumentos T&M), Trescal aporta su experiencia en la adquisición e implementación de sistemas de medición y soluciones de gestión de activos metrológicos. La empresa también ofrece formación y asesoramiento técnico. Trescal emplea a 3.900 personas en una red de más de 160 laboratorios de calibración. Trescal tiene una cartera de 70.000 clientes en las industrias de defensa, aeroespacial, aeronáutica, automotriz, energética, electrónica, telecomunicaciones, química y farmacéutica. Su facturación proforma es de 350 millones de euros. www.trescal.com.
Hands placing last piece of a Puzzle
Concepto Pharma 4.0. Evolución y tendencias de los sistemas informáticos, y transformación digital
La industria 4.0. brinda nuevas oportunidades para la industria Life Science, pero dichas tecnologías deben ser afrontadas con un modelo apropiado basado en el pensamiento crítico, en donde cada vez más el proveedor tecnológico juega un papel relevante.
Introducción
La Industria 4.0, que representa la cuarta revolución industrial, ha llegado a todos los ámbitos de la industria, y el sector farmacéutico y afín, no es una excepción. La presión por la innovación y la eficiencia está haciendo que las compañías farmacéuticas reconsideren la forma en que operan en todos sus ámbitos. Con motivo de la rápida evolución en la tecnología existente, varias tendencias están conduciendo a una digitalización y automatización tanto de los procesos productivos, de laboratorio, logística, gestión de calidad, así como los relacionados con el entorno de la investigación, ensayos clínicos y la Farmacovigilancia. Las tecnologías de Industria 4.0 están revolucionando y revolucionarán muchas áreas, y la forma en que gestionamos la calidad de nuestros datos.
Entre las tendencias y tecnologías clave de la Industria 4.0 se encuentran:
Big Data, Análisis predictivo, centros operativos conectados
La Industria 4.0 representa la era de la interconexión inteligente de los procesos (networking) para el mejor tratamiento de los datos y así ofrecer una mejor productividad, gestión de los recursos, optimización de los procesos y toma de decisiones, escalabilidad y flexibilidad en tiempo real de forma personalizada. Esta capacidad en el tratamiento de los datos para la mejora en continuo de nuestros procesos, marca la gran diferencia respecto a la automatización de los procesos que representaba la Industria 3.0 de los sistemas del pasado.
A medida que ha ido avanzando la tecnología, los procesos de negocio han ido cobrando mayor complejidad, con una mayor presencia de herramientas digitales avanzadas que permiten romper comportamientos o usos no adecuados con el foco en la mejora en continuo de nuestros procesos, eliminar errores humanos, reducir costes y retrasos, y garantizar la conformidad reguladora de los procesos, sistemas y por lo tanto de los productos.
En las últimas décadas hemos ido pasando de procesos manuales basados íntegramente en papel, a procesos digitales con registros electrónicos y firmas electrónicas, sistemas de computación en la nube a, por último, sistemas inteligentes con capacidad de análisis predictivos para la toma de decisiones en tiempo real.
Imagen 2
¿Qué se requiere de los sistemas informáticos?
Los sistemas informáticos actuales deben ser conformes al entorno regulador GxP y de Data Integrity, y deben ser escalables, flexibles, así como facilitar nuestro trabajo, optimizando flujos, y recursos, para que el tratamiento de la información reguladora y la toma de decisiones esté basada en información objetiva y veraz, habiendo eliminado del proceso de decisión, la incertidumbre asociada. Para ello, la aplicación de medidas de control a través del uso del conocimiento, así como la implantación de medidas organizativas son puntos relevantes.
Por todo lo anterior, a la hora de elegir nuestro sistema informático, debemos valorar que éste disponga o cumpla con los siguientes requisitos:
Comprensivo, ágil, intuitivo: Interfaz de usuario fácil e intuitiva adaptable a los flujos del proceso. Búsqueda y consulta ágil de la información.
Facilitador de la toma de decisión basada en el riesgo.
Dispuesto con conexión global (datos, procesos y personas).
Visión holística de los procesos y flujos.
Conectado con otros sistemas informáticos en tiempo real: Integridad ágil y efectiva con otros sistemas informáticos (ej.: ERP, LIMS, etc.), así como infraestructura IT (ej.: BBDD, Directorio activo, servidor de correo, Office, y buscadores de información).
Herramientas avanzadas de predicción de datos (ej.: AI, ML).
Comportamiento predictivo.
Escalable, flexible, dinámico, inteligente: Adaptación modular, extensible al entorno. Funciones analíticas y de reporte. Datos actualizados en tiempo real.
Mejora de los flujos de trabajo. Catalizador de mejoras (promotor de la mejora continua)
A su vez, nuestro sistema informático debe estar diseñado, desarrollado y mantenido dentro de un sistema de gestión de calidad (SGC), por lo que es importante incluir los siguientes requisitos:
Experimentado dentro de un SGC: Historia del producto/solución dentro del sector.
Desarrollado e implementado bajo un modelo comprensible (GAMP5).
Validación: Solución Validada. Paquete de validación, y mantenimiento del estado de control.
Seguro: Medidas de seguridad de protección de datos para asegurar su integridad y privacidad. Entorno web seguro (en el caso de sistemas XaaS[1]).
Conforme desde el inicio: Marco regulador GxP, Data Integrity (incluyendo 21 CFR Part 11, Anexo 11 EU GMP).
Es importante reconocer además que la digitalización y las tecnologías emergentes en la fabricación y el control pueden presentar ciertos desafíos. Por ello, cabe destacar la importancia de la aplicación del QRM (Quality Risk Managament) en el establecimiento de sus requisitos y diseño, teniendo en consideración la transferencia de tecnología de los procesos de producción avanzados, los métodos analíticos, así como en los métodos avanzados de análisis de datos durante todo el ciclo de vida de nuestros sistemas informáticos.
Nuevo enfoque de calidad y relevancia del proveedor tecnológico
El marco regulador existente está siendo revisado en la actualidad (ej.: segunda edición de la GAMP5, nuevo enfoque CSA[2] de la FDA, nueva revisión de la ICH Q9 (R1)) para abordar la creciente importancia de los proveedores de servicios, los enfoques en el desarrollo de software y el uso más generalizado de nuevas tecnologías, herramientas de software y automatización. El uso del pensamiento crítico con los conocimientos y la experiencia para definir los enfoques apropiados son puntos relevantes a tener en cuenta.
Las nuevas tecnologías de AI/ML requieren además de modelos para determinar la estrategia y nivel de esfuerzo de las actividades de validación para poder proporcionar una justificación para la distinción entre los niveles de validación en función de la autonomía y diseño de control del modelo de IA.
Guías de la ISPE como, la GAMP® RDI Good Practice Guide: Data Integrity by Design, la GAMP® Good Practice Guide: Enabling Innovation – Critical Thinking, Agile, IT Service Management, o la GAMP® Good Practice Guide: IT Infrastructure Control and Compliance(Second Edition) intentan clarificar y dotar a la industria de escenarios de referencia relativos a las implicaciones de las diversas tecnologías de la industria 4.0 cada vez más comunes, para el correcto ciclo de vida tanto de los sistemas informáticos, infraestructura IT y/o de los datos GxP.
El nuevo enfoque de calidad pretende aclarar la interpretación de los requisitos regulatorios con el entorno tecnológico actual, para cambiar el foco de la documentación al pensamiento crítico durante todo el ciclo de vida del sistema informático y de su infraestructura de IT que lo soporta.
El objetivo, con este nuevo marco normativo, es ayudar a las empresas reguladas a facilitar el mejor uso de los recursos y fomentar la aplicación de prácticas adecuadas y proporcionadas a QRM, la calidad del producto, la seguridad del paciente y la integridad de los datos.
Imagen 3
El papel del proveedor tecnológico
Cuánto más complejo sea nuestro sistema informático, y mayor impacto posea en nuestras funciones de negocio, mayor relevancia cobra nuestro proveedor para el éxito de nuestro proyecto de IT. Por ello, es importante tener en consideración los siguientes puntos en la inter-relación con nuestro proveedor de IT y su papel como experto en la materia (SME):
Aprovechamiento del proveedor como experto para el incremento del cumplimiento con las perspectivas reguladoras mediante definición de un modelo de ciclo de vida experimentado: Pensamiento crítico para determinar tecnología, metodología y estrategia (GAMP5, Agile, IT Service Management).
Seguimiento de estándares y marco regulador GxP, Data Integrity, ISO, Buenas Prácticas Documentales, GEP (Good Engineering Practices), Sistema de gestión de calidad IT, seguridad de la información, ITIL (Information Technology Infrastructure Library).
Acompañamiento del proveedor en la definición de requisitos facilitando mejoras, automatización de procesos y optimización de tareas para mayor agilidad y eficiencia. Actividades coordinadas iniciadas tempranamente (espíritu de equipo cliente/proveedor).
Cooperación interfuncional y efectiva entre el lado técnico y calidad.
Optimización de tiempos y recursos para el logro de la conformidad: Reducción de costes y tiempos necesarios para conseguir un sistema conforme. Evitar errores.
Resolución de dudas y guía durante todo el proceso que implicará en el cliente en un mayor control del proceso.
Generación de las especificaciones del sistema: Especificaciones de configuración (CS, Configuration Specification), Especificaciones Funcionales (FS, Functional Specification).
Provisión del paquete de verificación/validación: Informes de instalación y configuración y puesta en servicio, paquete de pruebas de validación según modelo de servicio requerido y acordado (generación de las evidencias documentales de soporte de valor añadido a la calidad de las pruebas para la verificación activa de búsqueda de defectos).
Soporte en la gestión de los cambios, configuraciones, problemas, servicios de IT según responsabilidad del modelo de servicio contratado.
Soporte continuado del proveedor en formaciones.
Por todo lo anterior, la competencia y la fiabilidad del proveedor son factores claves a la hora de seleccionar un producto o proveedor de servicios. Por ello, es importante realizar una evaluación con estándares adecuados del proveedor de SW o de servicios IT, ya sea para suministrar, instalar, configurar, integrar, validar, mantener, modificar o conservar un sistema informático o un servicio de IT relacionado. Por ello, debemos contar con procedimientos específicos de evaluación de proveedores tecnológicos que nos permitan conocer si dicho proveedor será o no capaz de cumplir con nuestros requisitos de negocio, reguladores y de integridad de datos. Dichos proveedores deben garantizar la validación continua del software y/o de cualificación de la infraestructura IT relativa, según corresponda, según los acuerdos establecidos[3].
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de infraestructura IT. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución de su proyecto de digitalización desde el punto de vista de calidad, y asegurar el ciclo de vida de su sistema informático y/o infraestructura IT, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros.
[1] XaaS: “X” as a Service. “X” como un Servicio, donde “X” puede ser: “Platform” PaaS (Platform as a Service); IaaS (Infrastructure as a Service); SaaS (Software as a Service).
[2] Guía borrador “CSA Computer Software Assurance for Production and Quality System Software” del centro CDRH (Center for Devices and Radiological Health).
[3] Los departamentos de informática (IT) deben considerarse análogamente a los proveedores de IT, en caso que nuestro sistema informático sea desarrollado y/o mantenido internamente.
Hands placing last piece of a Puzzle
La FDA publica su borrador guía sobre CSA “Computer Software Assurance” para revisión a comentarios
En los últimos años, los avances tecnológicos en los procesos de fabricación, incluyendo la automatización, la digitalización, la robótica, así como los entornos y metodologías de test, han permitido a los fabricantes reducir fuentes de error, optimizar recursos, y reducir el riesgo en la salud del paciente. La FDA reconoce el potencial de estas tecnologías para brindar beneficios significativos para mejorar la calidad, la disponibilidad y la seguridad de los dispositivos médicos, y ha emprendido varios esfuerzos para ayudar a fomentar la adopción y el uso de dichas tecnologías. Dentro de este marco, se publica el nuevo borrador guía como referente para el sector Life Science sobre el nuevo enfoque ”Computer Sofware Assurance”.
Nuevo enfoque para la garantía de calidad del software
El nuevo borrador guía sobre Validación del software “Computer Software Assurance for Production and Quality System Software” recomienda que la «garantía de la calidad del software» se centre en prevenir la introducción de defectos en el proceso de desarrollo de software y fomenta el uso de un enfoque basado en el riesgo para establecer la confianza conforme el software es adecuado para su uso previsto.
Con esta guía la FDA pretende proporcionar recomendaciones sobre la garantía del software informático involucrado en los procesos de producción o del sistema de calidad de un dispositivo médico. Esta guía tiene por objeto:
Describir la «garantía del software informático» como un enfoque basado en el riesgo para establecer la confianza en la automatización utilizada para los sistemas de producción o calidad, e identificar dónde puede ser apropiado un rigor adicional.
Describir diferentes métodos y actividades de test aplicables para establecer la garantía del software informático y proporcionar así evidencia objetiva para cumplir con los requisitos reguladores.
Periodo de consulta
Se establece un periodo de consulta para el envío de comentarios y sugerencias de 60 días a la publicación en el Registro Federal del aviso de disponibilidad de la guía preliminar, por lo que se pueden enviar comentarios y sugerencias hasta el 13 de noviembre 2022.
Aproximación basada en el riesgo
En el borrador guía, la FDA describe un marco de riesgo de aseguramiento del software que incluye la evaluación del uso previsto del sistema informático basado en el riesgo para identificar las actividades de aseguramiento correctas basadas en su nivel de riesgo y crear un registro para documentar que el sistema funcione según lo previsto.
La FDA hace una distinción en la guía entre el software que es parte directa del proceso de producción y el software que respalda el sistema de calidad. Se reconoce además que el software puede tener más de un uso previsto, lo que puede conllevar diferentes riesgos según sus características, funciones y operaciones dentro del proceso de producción o del sistema de calidad.
“FDA recommends that manufacturers document their decision-making process for determining whether a software feature, function, or operation is intended for use as part of production or the quality system in their Standard Operating Procedures (SOPs)”.
Una vez el software ha sido clasificado debe emplearse un análisis basado en el riesgo para identificar las actividades de aseguramiento apropiadas para el software. Todo ello, incluye la identificación de peligros que pudieran impactar en el funcionamiento previsto del sistema y por lo tanto en la producción o el sistema de calidad (riesgo de proceso) o causar un daño en el paciente (seguridad).
“A feature, function, or operation that could lead to severe harm to a patient or user would generally be high device risk. In contrast, a feature, function, or operation that would not foreseeably lead to severe harm would likely not be high device risk.”
Entre las actividades de aseguramiento a aplicar, las pruebas “Scripted Testing” con instrucciones previas escritas de casos de test (como son los test tradicionales de una validación) pueden ser más apropiadas para los sistemas de mayor riesgo, al ser este tipo de pruebas de mayor rigor, mientras que las pruebas “Unscripted Testing” sin instrucciones previas escritas de casos de test y de menor carga documental (tales como pruebas ad-hoc, adivinación de errores, pruebas exploratorias o una combinación de las mismas) pueden ser más apropiadas para sistemas de menor riesgo (pruebas menos planeadas, y limitadas). En este sentido, las pruebas “Unscripted Testing” pueden ser las precursoras de las “Scripted Testing” más formales y rigurosas de estadios de prueba más maduros y formales dentro del proceso de desarrollo y verificación del software.
Los fabricantes también deben asegurarse de crear un registro apropiado para documentar que la característica, función u operación del software funciona según lo previsto. El registro apropiado debe incluir el uso previsto, la determinación de los riesgos y la documentación de las actividades de aseguramiento a realizar. Sobre este asunto, la nueva guía pretende crear una nueva oportunidad para racionalizar la documentación, al aplicar el pensamiento crítico y la gestión de riesgos con relación al impacto del sistema en la seguridad del paciente y el producto dentro de un enfoque de garantía de calidad.
“Documentation of assurance activities need not include more evidence than necessary to show that the software feature, function, or operation performs as intended for the risk identified. FDA recommends the record retain sufficient details of the assurance activity to serve as a baseline for improvements or as a reference point if issues occur”.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, validación de sistemas, cualificación de equipos y calibración. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten:
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Proceso de desarrollo y mantenimiento del software de dispositivo médico.
Evaluaciones de Sistemas informáticos y Data Integrity: verificación 21 CFR Part 11, Data Integrity Risk Assessment (DIRA), data process mapping, data integrity remediation plan, orphan data check).
Desarrollo del VMP / VP.
Revisión y evaluación de metodologías y actividades de test. GAP análisis, AMFE.
Desarrollo de procedimientos normalizados de trabajo.
Desarrollo de los expedientes de validación.
Pruebas de aceptación FAT / SAT / Comisionado.
Cualificacion de Infraestructura IT (QP, RA, DQ, IQ, OQ, PQ, gestión de cambios y revisión periódica).
Evaluación de proveedores de SW / tecnológicos.
Acompañamiento a proveedores en pruebas de aceptación.
Preparación de procedimientos de gestión y mantenimiento del sistema informático.
Implantación de Sistemas de Calidad IT. Sistemas de Calidad para desarrolladores de software.
Reevaluaciones periódicas.
Procesos de migración de datos.
Evaluación ante la retirada de un sistema informático.
Plan de Transformación CSA.
Actividades de formación dirigida.
Si está evaluando la necesidad de contar con un proveedor experto que le acompañe en este proceso de transformación al nuevo enfoque CSA desde la órbita de calidad, y /o asegurar y agilizar la correcta consecución del ciclo de vida de su sistema informático y/o infraestructura IT, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
iStock_000020268060Large
Nuevo anexo 1 EU GMP y estrategia de control de la contaminación (CCS). ¿Estás preparado?
El entorno regulador y tecnológico en la fabricación de medicamentos estériles avanza, y por ello, es importante estar preparado y contar con proveedores expertos que acompañen a los fabricantes en este proceso de transformación. El control de la contaminación y la elaboración de una estrategia de control de la contaminación es un concepto relevante que define la nueva versión del Anexo 1.
Estrategia de control de la contaminación
El Anexo 1 GMP de la EU sobre «Fabricación de medicamentos estériles« ha sido revisado a finales de agosto tras varios años de ansiada espera. Tal y como mencionábamos en una anterior noticia, la nueva versión hace foco en una mejor aplicación del proceso de gestión de los riesgos (ICH Q9 – Quality Risk Management) y del sistema de calidad farmacéutico (ICH Q10 – Pharmaceutical Quality System) para asegurar un mayor control de la contaminación. En este ámbito, el nuevo anexo enfatiza la implementación de una estrategia de control de la contaminación (CCS, Contamination Control Strategy, por sus siglas en inglés) para definir los puntos críticos de control y evaluar la eficacia de todos los controles (de diseño, procedimiento, técnicos y organizativos), así como las medidas de seguimiento empleadas.
“Contamination Control Strategy (CCS) – A planned set of controls for microorganisms, endotoxin/pyrogen and particles, derived from current product and process understanding that assures process performance and product quality. The controls can include parameters and attributes related to active substance, excipient and drug product materials and components, facility and equipment operating conditions, in-process controls, finished product specifications, and the associated methods and frequency of monitoring and control.”(Glossary)
El nivel de esfuerzo y de detalle de la estrategia de control de la contaminación debe ser proporcional al tipo de proceso y producto. Debe considerarse que el nuevo Anexo 1 no solo provee directriz sobre la fabricación de productos «estériles» sino que también brinda orientación sobre la fabricación de los productos “no estériles”:
“The intent of the Annex is to provide guidance for the manufacture of sterile products. However, some of the principles and guidance, such as contamination control strategy, design of premises, cleanroom classification, qualification, validation, monitoring and personnel gowning, may be used to support the manufacture of other products that are not intended to be sterile such as certain liquids, creams, ointments and low bioburden biological intermediates, but where the control and reduction of microbial, particulate and endotoxin/pyrogen contamination is considered important. Where a manufacturer elects to apply guidance herein to non-sterile products, the manufacturer should clearly document which principles have been applied and acknowledge that compliance with those principles should be demonstrated.” (1 Scope)
Por otro lado, la estrategia de control de la contaminación debe revisarse activamente y, ser actualizada correspondientemente para impulsar la mejora continua de los métodos de fabricación y control. Su eficacia debe formar parte de la gestión de los procesos de revisión periódica.
Elementos a considerar dentro de la estrategia de control de contaminación
Tal y como menciona el Anexo 1, los elementos a considerar dentro de un CCS deben ser (pero no limitados a éstos) los siguientes:
Diseño de la planta y de los procesos, incluyendo la documentación asociada.
Locales y equipos.
Personal.
Servicios.
Controles de materias primas, incluyendo los controles en proceso.
Envases de productos y cierres.
Aprobación de proveedores.
Gestión de actividades subcontratadas y disponibilidad/transferencia de la información crítica entre las partes (ej.: servicios de esterilización).
Proceso de gestión de riesgos.
Validación del proceso.
Validación de procesos de esterilización.
Mantenimiento preventivo: mantenimiento de equipos, servicios e instalaciones (mantenimiento planificado y no planificado) para asegurar que no haya riesgo adicional de contaminación.
Limpieza y desinfección.
Sistemas de monitorización, incluyendo una evaluación de la viabilidad de la introducción de métodos alternativos científicamente sólidos que optimicen la detección de la contaminación ambiental.
Mecanismos de prevención: análisis de tendencias, investigación detallada, determinación de la causa raíz, acciones correctivas y preventivas (CAPA) y la necesidad de herramientas de investigación exhaustivas.
Mejora continua en base a la información derivada de lo anterior.
Desarrollo, documentación y mantenimiento de la estrategia de control de la contaminación
Los fabricantes de productos farmacéuticos, así como sus proveedores son conscientes que la contaminación puede provocar pérdidas de producto, o pueden representar un riesgo importante sobre la protección de la salud pública.
Es por ello crucial, la preparación de un documento CCS que brinde una descripción general de la totalidad de las medidas de control de la contaminación y su vínculo con la estrategia general CSS. El proceso de implantación de la estrategia de control de la contaminación se puede resumir en las siguientes etapas:
Etapa 1: Desarrollo (o revisión/mejora) de la estrategia de control de la contaminación (ej.: revisión de requisitos y necesidades, desarrollo de un Gap análisis para la detección de vulnerabilidades y su corrección).
Estado 2: Compilación de los documentos CCS (documentar todas las medidas, incluyendo procedimientos, QRM, controles, reportes de cualificación y validación, etc.) para el desarrollo del documento CSS.
Etapa 3: Evaluación y mantenimiento del CCS (revisiones en continuo, revisiones periódicas).
Tal y como especifica el propio Anexo 1, el desarrollo del CCS requiere de conocimiento técnico y de proceso detallado. El fabricante debe poseer suficiente conocimiento y experiencia sobre los productos fabricados, los equipos y los métodos de fabricación e ingeniería empleados para evaluar su impacto en la calidad del producto.
“The development of the CCS requires detailed technical and process knowledge. Potential sources of contamination are attributable to microbial and cellular debris (e.g. pyrogen, endotoxin) as well as particulate (e.g. glass and other visible and sub-visible particles).” (2 Principle)
Cobra relevancia el soporte de los proveedores y su correcta evaluación previa como expertos en la materia para el correcto acompañamiento durante todo el ciclo de vida de los sistemas y equipos clave. Ej.: proveedores de equipos y componentes críticos, esterilización, desinfectantes y detergentes, bioindicadores, ingenierías de servicios farmacéuticos (agua, vapor, gases), mantenimiento, consultoría y cualificación.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, validación de sistemas, cualificación de equipos y calibración. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten:
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Gap análisis de la estrategia actual de control de la contaminación.
Evaluación de proveedores.
Cualificación de salas limpias.
Cualificación de servicios farmacéuticos.
Cualificación de equipos de esterilización.
Establecimiento y mantenimiento de los planes de verificación de salas limpias.
Desarrollo del plan de estrategia de control de la contaminación.
Validaciones de proceso.
Validaciones de limpieza.
Desarrollo de los expedientes de validación / cualificación.
Acompañamiento a proveedores en pruebas de aceptación.
Revisiones periódicas.
Calibración instrumental.
Actividades de formación dirigida.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta implantación de su estrategia de control de contaminación, así como la correcta validación/cualificación y estado de control de sus instalaciones/equipos desde la órbita de calidad, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
GMP Qualification and validation
PUBLICAÇÃO DO ANEXO 1 EU GMP: ACTUALIZAÇÃO DO NOVO AMBIENTE REGULAMENTAR E DE FABRICO
Introdução e base para a nova revisão
A fim de atualizar o atual ambiente regulamentar e tecnológico no fabrico de medicamentos esterilizados, o Anexo 1 foi revisto. Após quase 5 anos de espera desde o primeiro projeto em 2017 e mais 2 anos desde o segundo projeto em 2020 com a revisão dos respetivos comentários públicos, a Comissão da UE publicou em 22 de Agosto o tão esperado Anexo 1 das GMP da UE sobre «Fabricação de medicamentos esterilizados».
O novo anexo esclarece como as empresas fabricantes podem tirar partido das novas possibilidades resultantes da implementação de uma melhor compreensão do processo de gestão de riscos (ICH Q9 – Gestão de Riscos de Qualidade) e do sistema de qualidade farmacêutica (ICH Q10 – Sistema de Qualidade Farmacêutica). Inclui também novos desenvolvimentos tecnológicos que exigiram a revisão do Anexo 1. Por exemplo, nova secção sobre Estratégia de Controlo de Contaminação (CCS), e novas secções para acomodar os recentes avanços na tecnologia de fabrico estéril, tais como Sistemas de Barreiras de Acesso Restrito (RABS) e isoladores.
A revisão do Anexo 1 também tem em conta as alterações noutros capítulos e anexos de GMP e/ou liga-os para controlo posterior (por exemplo, Capítulo 1 sobre Sistema de Qualidade Farmacêutico, Anexo 4 sobre Fabrico de medicamentos veterinários que não medicamentos veterinários imunológicos, Anexo 12 sobre Utilização de Radiações Ionizantes no fabrico de medicamentos, Anexo 15 sobre Qualificação e Validação, Anexo 17 sobre Libertação Paramétrica), bem como outros regulamentos relacionados (ISO 14644).
Principais alterações em relação ao projeto para 2020
Em comparação com o projeto anterior de 2020, a estrutura básica do Anexo 1 permaneceu inalterada, mas a nova revisão é mais abrangente, aumentando de 52 para 58 páginas, e há numerosas supressões, resumos e novas inserções em muitos capítulos.
Por exemplo, o subcapítulo «Tecnologias de Barreiras» no Capítulo 4 «Instalações» é quase duplicado para tratar em maior detalhe a utilização de luvas e materiais, bem como métodos de descontaminação em RABS e isoladores separadamente, e os subcapítulos «Form-Fill-SEAL (FFS)» e «Blow-Seleal» (equipamento utilizado no fabrico de produtos terminados esterilizados) no Capítulo 8 «Produção e Tecnologias Específicas» quase triplicam em conteúdo.
Prazos
O novo Anexo 1 entrará em vigor a 25 de Agosto de 2023, ou seja, um ano após a publicação no Eudralex Volume 4, com exceção do ponto 8.123 (esterilização em liofilizadores), que será aplicável a 25 de Agosto de 2024.
Trescal
TRESCAL é um prestador de serviços global para a indústria das Ciências da Vida. Oferece serviços de implementação/consultoria de qualidade, validação de sistemas, qualificação de equipamentos e calibração. O nosso conhecimento específico e transversal permite-nos oferecer um serviço de qualidade global como especialistas na área, onde quer que necessite.
Se estiver a avaliar a necessidade de um fornecedor especializado para assegurar e acelerar a correta realização e validação do seu processo de esterilização, ou para resolver com sucesso o seu projeto de qualificação de sala limpa e/ou equipamento de esterilização a partir da órbita da qualidade, poupando eventos e recursos imprevistos, não perca mais tempo e contacte-nos, teremos todo o prazer em ouvi-lo e juntar-se ao seu projeto.
GMP Qualification and validation
Publicación del anexo 1 EU GMP: Actualización al nuevo entorno regulador y de fabricación
Introducción y bases de la nueva revisión
Con motivo de actualizar el entorno actual regulador y tecnológico en la fabricación de medicamentos estériles, el Anexo 1 ha sido revisado. Tras casi 5 años de espera desde el primer borrador en el año 2017 y más 2 años desde el segundo borrador en el año 2020 con la revisión de sus respectivos comentarios públicos, la Comisión de la EU ha publicado el pasado 22 de agosto, el esperado Anexo 1 GMP de la EU sobre «Fabricación de medicamentos estériles«.
El nuevo anexo clarifica cómo las empresas fabricantes pueden aprovechar las nuevas posibilidades derivadas de la aplicación de una comprensión mejorada del proceso de gestión de los riesgos (ICH Q9 – Quality Risk Management) y del sistema de calidad farmacéutico (ICH Q10 – Pharmaceutical Quality System). También incluye nuevos desarrollos tecnológicos que han hecho necesaria la revisión del Anexo 1. Ej.: nueva sección sobre estrategia de control de la contaminación (CCS, Contamination Control Strategy, por sus siglas en inglés), y nuevas secciones para adoptar los recientes avances en la tecnología de fabricación estéril como son los sistemas de barrera con acceso restringido (RABS, Restricted Access Barrier System, por sus siglas en inglés) y los aisladores.
La revisión del Anexo 1 tiene en cuenta, además, los cambios en otros capítulos y anexos GMP y/o los relaciona para mayor control (ej.: Capítulo 1 sobre Sistema de Calidad Farmacéutico, anexo 4 sobre Fabricación de medicamentos veterinarios distintos de los medicamentos veterinarios inmunológicos, anexo 12 sobre Uso de las radiaciones ionizantes en la fabricación de medicamentos, anexo 15 sobre Cualificación y Validación, anexo 17 sobre Liberación Paramétrica), así como otras regulaciones relacionadas (ISO 14644).
Principales cambios respecto el borrador del año 2020
Comparativamente con el anterior borrador del año 2020, la estructura básica del Anexo 1 se ha mantenido sin cambios, pero la nueva revisión es más completa, pasando de 52 a 58 páginas, y existen numerosas deleciones, resúmenes y nuevas inserciones en numerosos capítulos.
Por ejemplo, el subcapítulo «Barrier Technologies» en el capítulo 4 «Premises» casi se duplica para tratar con mayor detalle el uso de guantes y materiales, así como de métodos de descontaminación en RABS y aisladores por separado, y los subcapítulos «Form-Fill-SEAL (FFS)» y «Blow-Seleal» (equipos utilizados en la fabricación de productos con esterilización terminal) en el capítulo 8 «Production and Specific Technologies» casi triplican su contenido.
Plazos
El nuevo Anexo 1 entrará en vigor el 25 de agosto de 2023, es decir, un año después de la publicación en Eudralex Volumen 4, a excepción del punto 8.123 (esterilización en liofilizadores), que será de aplicación el 25 de agosto de 2024.
Trescal
TRESCALes un proveedor global de servicios para la industria Life Science. Ofrece servicios de implantación/consultoría de calidad, validación de sistemas, cualificación de equipos y calibración. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución y validación de su proceso de esterilización, o afrontar con éxito su proyecto de cualificación de sala limpia y/o equipo de esterilización desde la órbita de calidad, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Hands placing last piece of a Puzzle
Segunda Edición Guía ISPE GAMP® 5: Actualización a los nuevos modelos, requisitos y usos tecnológicos
Introducción y bases de la segunda edición
Tras 14 años de la primera edición de la Guía ISPE GAMP® 5 A Risk-Based Approach to Compliant GxP Computerized Systems, este mes de julio se ha publicado finalmente su segunda y esperada edición.
Las guías GAMP pretenden ofrecer un marco de referencia en buenas prácticas dentro del sector Life Science para garantizar que los sistemas informáticos sean efectivos, confiables y de alta calidad. De esta forma son adecuados para su uso previsto y cumplen con las regulaciones aplicables.
Manteniendo los principios y el marco de la primera edición, la nueva guía ISPE GAMP® 5 actualiza su aplicación habiendo revisado las prácticas, regulaciones y desarrollos tecnológicos actuales, para incluir la mayor relevancia de los proveedores IT de servicios, los enfoques en evolución del desarrollo de software, y el uso ampliado de herramientas de software y automatización. Destaca el uso del pensamiento crítico de los expertos en la materia (SME, subject matter expert por sus siglas en inglés) para poder definir los enfoques más apropiados en las circunstancias específicas en base a su conocimiento y experiencia.
Por lo tanto, la nueva guía ISPE GAMP® 5 incluye y fomenta lo siguiente:
Mayor relevancia de los proveedores de servicios de IT. Incluyendo los proveedores de servicios en la nube (cloud), para alentar a las empresas reguladas a maximizar la participación de los proveedores y así aprovechar su conocimiento, experiencia y documentación, cuando sea posible.
Enfoques en evolución del desarrollo de software, enfatizando que el enfoque de especificación y verificación GAMP no es inherentemente lineal, sino que también admite perfectamente métodos iterativos e incrementales.
Mayor uso de herramientas de software y automatización para lograr un mayor control, calidad y menores riesgos durante todo el ciclo de vida del sistema informático.
Además, la nueva guía, incluye el nuevo enfoque Computer Software Assurance (CSA, por sus siglas en inglés) de la FDA, y clarifica materias referenciando otras guías GAMP RDI sobre integridad de datos (DI, Data Integrity, por sus siglas en inglés). Además, incluye nuevas consideraciones sobre el uso de software open-source (OOS, Open Source Software, por sus siglas en inglés).
Mientras que los enfoques de especificación y verificación, así como el marco general y conceptos clave del ciclo de vida del sistema, y el enfoque de gestión de riesgos de calidad (alineado con la ICH Q9) permanecen sin cambios, el contenido técnico de la guía ha sido revisado. Por ello, los apéndices de la nueva guía han sido actualizados para incorporar las prácticas, requisitos regulatorios y tecnologías actuales, dentro de la evolución tecnológica de la industria del sector Life Science y el marco Pharma 4.0TM.
Por ello, son de nueva creación los siguientes apéndices: D8 (Agile). D9 (Software Tools). D10 (Blockchain). D11 (Artificial Intelligence and Machine Learning – AI/ML, por sus siglas en inglés-). M11 (IT Infrastructure) y M12 (Critical Thinking).
Además, han sido significativamente revisados los siguientes apéndices: D1 (Specifying Requirements), y S2 (Electronic Production Records).
Por el contrario, han sido eliminados los siguientes apéndices: D2 (Functional Specifications) que ha sido combinado junto el apéndice D1 para estar en un único apéndice, O7 (Repair Activity) que ha sido revisado para estar definido en el apéndice O6 (Operational Change and Configuration Management) y S5 (Managing Quality within an Outsourced IS/IT Enviromnment) cuyo contenido ha sido revisado para ser incluido dentro del nuevo apéndice M11 (IT Infrastructure).
A su vez, la nueva guía considera que son áreas sinérgicas, tanto la Gestión del conocimiento, así como el Advancing Pharmaceutical Quality (APQ, por sus siglas en inglés) y la transformación digital Pharma 4.0 TM, siendo la madurez digital y la integridad de datos elementos facilitadores para el establecimiento de una efectiva estrategia de transformación digital.
¿A quién está dirigida?
La nueva guía está destinada tanto a empresas reguladas, como proveedores y reguladores. Es importante aclarar que, por proveedores, se incluyen a proveedores de software, hardware, equipos, servicios de integración de sistemas, servicios IT, tanto internos como externos a la empresa regulada.
Trescal
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de infraestructura IT. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia, en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del ciclo de vida de su sistema informático, infraestructura IT, o afrontar con éxito su proyecto de transformación digital desde la órbita de calidad, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
iStock_000016752326Large
UNE-EN ISO 13485:2018
Desde Abril 2022 Trescal dispone de la certificación en ISO13485:2016.
Con el siguiente alcance:
Calibración
Inspección
Mantenimiento
Reparación de equipos electromédicos
Como decisión estratégica se ha optado por adquirir la certificación bajo la norma ISO 13485:2016 (UNE-EN ISO 13485:2018). Productos sanitarios. Sistemas de gestión de la calidad. Requisitos reglamentarios. Eligiendo a ASSI Trescal que sita en Barcelona, como laboratorio poseedor de esta certificación, por su gran actividad en el sector biomédico.
Esta norma establece los requisitos para un sistema de gestión de la calidad de las organizaciones involucradas en las diferentes etapas del ciclo de vida de un producto sanitario. Abarca tanto a fabricantes de estos productos como a proveedores. En el caso de ASSI Trescal, como empresa de servicios de calibración y mantenimiento, se ha elegido voluntariamente cumplir con estos requisitos y certificarnos.
Ha sido muy fácil integrar los requisitos de la norma a nuestro Sistema de Calidad. ASSI Trescal posee un gran conocimiento en el sector al que nos dirigimos y sabe perfectamente cuáles son los requerimientos de sus clientes y los reglamentos aplicables.
Cualificación de la infraestructura IT y estrategia de control en su ciclo de vida.
A medida que las organizaciones han ido dependiendo de los sistemas informáticos para la automatización de sus procesos y actividades reguladas, la infraestructura IT que los soporta ha ido cobrando cada vez mayor relevancia.
Situación actual y cualificación infraestructura IT
En los últimos años ha habido un salto significativo en el ámbito de la Infraestructura de la Tecnología de la Información (IT) que ha dado lugar a una reorientación y actualización de las políticas de validación/cualificación en esta materia. Aunque el anexo 11 de las EU GMP ya hacía alusión a este tema en su principio “La aplicación debe validarse; la infraestructura informatizada (IT) debe cualificarse”, la complejidad tecnológica y los problemas en Data Integrity, han derivado en un mayor aseguramiento del estado de control de la infraestructura IT.
El salto significativo en la Infraestructura IT se resume en los siguientes cambios del modelo tecnológico, muy bien recogidos en la Guía GAMP sobre IT Infrastructure Control and Compliance (agosto 2017):
El uso de tecnologías de virtualización que permiten compartir, combinar y maximizar los recursos.
El uso de sistemas Cloud, incluyendo las aplicaciones GxP «as-a-service» y los modelos de servicios de tipo XaaS: infraestructura como servicio (IaaS), plataforma como servicio (PaaS) y software como servicio (SaaS).
La mayor externalización de actividades IT en proveedores tecnológicos (Outsourcing) y uso de Third party data centers.
Gestión de los riesgos
La gestión de riesgos proporciona un método para identificar aspectos de una manera controlada y sistemática, y debe realizarse para cada sistema y/o proceso de infraestructura relevante. Durante este proceso, es importante el poder dar respuesta a las siguientes preguntas:
¿Qué aplicaciones GxP disponemos?
¿Cuál es la infraestructura IT que soporta estas aplicaciones GxP? ¿Qué plataformas, sistemas y/o componentes IT componen esta infraestructura IT? ¿Cuál es el estado de validación y/o cualificación de dichas plataformas, componentes? ¿Esta cualificación se encuentra correctamente documentada y soportada por un ejercicio de gestión de los riesgos –trazabilidad con los riesgos?
¿Se requieren controles adicionales para el adecuado control de los riesgos existentes incluyendo los riesgos en materia de Data Integrity? ¿Deben implantarte cambios?
Se realizan revisiones periódicas para la evaluación y adecuado control de la infraestructura IT?
Cualificación de la Infraestructura IT
La Cualificación de la Infraestructura IT es la confirmación mediante examen y provisión de evidencias objetivas que las especificaciones de la Infraestructura IT son conformes a los requerimientos del usuario y a su uso previsto, y que todos los requerimientos pueden ser consistentemente cumplidos.
Al tratarse, la Infraestructura IT, en su conjunto de una unidad compuesta por varios procesos, servicios, y plataformas compartidas, es importante abordar la estrategia de cualificación de forma segmentada, o global dependiendo del estudio de gestión de los riesgos previo para evitar retrabajos, y maximizar el esfuerzo de validación/cualificación en aquellas áreas de mayor riesgo.
Sistema de gestión de calidad y mantenimiento del estado cualificado
Es importante disponer de un sistema de gestión de calidad implantado y robusto que asegure el adecuado estado de control de la infraestructura IT. Por ello, es importante el revisar el estado actual de la documentación de nuestra infraestructura IT (Manual de Calidad con la inclusión de los procesos clave de infraestructura IT, Job Descriptions que definan la asignación de roles y responsabilidades, Procedimientos dirigidos al mantenimiento del estado de control y cualificación) por si fuera necesaria completarla y/o actualizarla, y el de rodearnos de personal experto en caso de necesitar ayuda o soporte para su adecuado desarrollo.
¿Qué ofrece Trescal?
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de infraestructura IT. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del ciclo de vida de su infraestructura IT, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
iStock_000019934130Large
Sistemas informáticos en la industria Life Science: Consultoría de calidad y Validación
Introducción y marco previo regulador
En los últimos años, la tecnología ha avanzado significativamente, y la industria regulada se ha apoyado en una gran variedad de sistemas informáticos para la realización de sus funciones de negocio. Además, el actual marco normativo se ha ido adaptando para dotar de mejor aseguramiento al estado de control de los sistemas informáticos durante todo su ciclo de vida, con especial foco en la calidad e integridad del dato.
Las guías de Data Integrity de los diferentes autoridades y agencias reguladoras, junto con el nuevo enfoque CSA de la FDA sobre Computer System Assurance, y la consecuente próxima publicación de la segunda edición de la Guía GAMP 5 tras 14 años de su primera edición, hace que el entorno de la industria Life Science ponga de nuevo el foco, aún más si cabe, en la gestión del ciclo de vida de los sistemas informáticos, así como en el ciclo de vida del dato.
Ciclo de vida de los sistemas informáticos
Disponer de un Sistema de Calidad IT es un elemento clave para asegurar el correcto ciclo de vida de nuestro sistema informático. Además. en un entorno cada vez más complejo y de mayor especialización técnica, la implicación del proveedor de la aplicación y/o tecnológico cobra mayor relevancia, por lo que es crucial su adecuada evaluación.
Las fases del ciclo de vida de un sistema informático según la Guía GAMP 5 son las siguientes:
Concepto: Establecimiento de los requerimientos, descripción general y consideraciones de diseño
Proyecto: Planificación, selección del proveedor y de la solución, especificaciones detalladas del sistema, y validación
Operación: Gestión del sistema en su fase operativa (ej.: operación, mantenimiento, gestión de cambios y configuración, revisiones periódicas, incidencias, copia de seguridad, restauración de datos, seguridad lógica y física, plan de contingencia).
Retirada: Retirada, migración, sustitución del sistema
Además, existen procesos o actividades de apoyo que tienen lugar en las fases del ciclo de vida que salen reforzadas inherentemente en el nuevo concepto CSA, como es la gestión de riesgos, la gestión del conocimiento, el pensamiento crítico, las buenas prácticas documentales, y/o la evaluación del proveedor.
Validación de sistemas informáticos
Según la FDA (21 CFR part 11), la validación de un sistema informático es la confirmación mediante examen y provisión de evidencias objetivas que las especificaciones de un sistema informatizado son conformes a los requerimientos del usuario y a su uso previsto, y que todos los requerimientos pueden ser consistentemente cumplidos.
Se trata pues de una actividad destinada a asegurar el uso previsto del sistema informático con el fin de cumplir con el marco regulador aplicable, así como conseguir las autorizaciones y certificaciones necesarias. Esta actividad además nos permite conocer mejor nuestro sistema, sus vulnerabilidades y oportunidades de mejora, no sólo en lo referente a aspectos de calidad, sino también de productividad. Aplicar una buena metodología en nuestro proyecto de validación permite una reducción del coste y tiempo necesarios para conseguir un sistema conforme, garantizando el cumplimiento de los requisitos y de las perspectivas reguladoras desde el inicio.
Ciclo de vida del dato
Con el nuevo enfoque Data Integrity, además de asegurar el correcto ciclo de vida del sistema informático, debemos asegurar el ciclo de vida del dato (principio ALCOA+).
Para ello, es crucial el poseer de un sistema maduro que incorpore la Gobernanza de los datos que abarque los siguientes conceptos clave.
Normativas de referencia de sistemas informáticos
Las empresas Life Science deben disponer de un sistema de gestión de calidad que asegure el correcto ciclo de vida de los sistemas informáticos y de sus datos conforme al marco regulador del producto en cuestión:
21 CFR Part 11: Criterios dirigidos por FDA para asegurar que los registros y firmas electrónicas son fiables y equivalentes a los registros y firmas escritas en papel.
Anexo 11 de las EU GMP: Requisitos de la comisión europea para los sistemas informáticos. Cuando un sistema informático reemplace una operación manual, no debe ser en detrimento de la calidad del producto, control del proceso o garantía de calidad. No debe haber un incremento del riesgo total del proceso.
Guía GAMP5: Guía desarrollada por la ISPE que promueve un ciclo de vida del sistema informático basado en buenas prácticas. Clarifica las responsabilidades y roles entre la industria farmacéutica y los proveedores de sistemas informáticos.
Guía GAMP IT Infrastructure Control and Compliance: Guía desarrollada por la ISPE que incluye la orientación para el control adecuado de la infraestructura informática (nuevas tecnologías de virtualización, sistemas/plataformas cloud / XaaS, seguridad, servidores, redes).
Data Integrity: Guías específicas de la FDA, MHRA, WHO, PIC/S, EMA, GAMP para asegurar la veracidad, consistencia, trazabilidad y disponibilidad de los datos durante todo el ciclo de vida de los datos.
Normas específicas de productos sanitarios:
ISO/TR 80002-2:2017 Medical device software – Part 2: Validation of software for medical device quality systems
UNE-EN 62304:2007/A1:2016 Software de dispositivos médicos. Procesos del ciclo de vida del software.
UNE-EN ISO 14971:2019 Dispositivos médicos/productos sanitarios. Aplicación de la gestión del riesgo a los MD.
UNE-EN 82304-1:2017 Software sanitario. Parte 1: Requisitos generales para la seguridad de los productos (Ratificada por la Asociación Española de Normalización en octubre de 2017.)
UNE-CEN ISO/TS 82304-2:2021 Software sanitario. Parte 2: Apps de salud y bienestar. Calidad y confiabilidad (ISO/TS 82304-2:2021) (Ratificada por la Asociación Española de Normalización en septiembre de 2021).
¿Qué ofrece Trescal?
Desde TRESCAL ofrecemos servicios de soporte relativos al ciclo de vida del sistema informático (sistema de gestión de calidad/producción), y/o del software de dispositivo médico según las normativas de aplicación, ofreciendo consultoría específica en las siguientes materias:
Establecimiento de los requerimientos técnicos y reguladores.
Revisión del diseño.
Proceso de desarrollo y mantenimiento del software de dispositivo médico.
Evaluaciones de Sistemas informáticos y Data Integrity: verificación 21 CFR Part 11, Data Integrity Risk Assessment (DIRA), data process mapping, GAP analysis, data integrity remediation plan, orphan data check).
Desarrollo del VMP / VP.
Establecimiento de estrategias.
Desarrollo de los expedientes de validación.
Pruebas de aceptación FAT / SAT / Comisionado.
Cualificacion de Infraestructura IT (QP, RA, DQ, IQ, OQ, PQ, gestión de cambios y revisión periódica).
Evaluación de proveedores de SW / tecnológicos.
Acompañamiento a proveedores en pruebas de aceptación.
Preparación de procedimientos de gestión y mantenimiento del sistema informático.
Implantación de Sistemas de Calidad IT. Sistemas de Calidad para desarrolladores de software.
Reevaluaciones periódicas.
Procesos de migración de datos.
Evaluación ante la retirada de un sistema informático.
Plan de Transformación CSA.
Actividades de formación dirigida.
TRESCAL es un proveedor global de servicios para la industria Life Science que ofrece servicios de implantación/consultoría de calidad, GxP, Quality Risk Management, Data Integrity, validación de sistemas y cualificación de infraestructura IT. Nuestro conocimiento específico y transversal nos permite ofrecer un servicio global de calidad como especialistas en la materia en los puntos que necesiten.
Si está evaluando la necesidad de contar con un proveedor experto para asegurar y agilizar la correcta consecución del ciclo de vida de su sistema informático y/o infraestructura IT, ahorrando imprevistos y recursos, no pierda más tiempo y contacte con nosotros, nos encantará tener noticias suyas y sumarnos a su proyecto.
Este navegador no es compatible con Trescal. Para asegurarse de que puede utilizar todas las funciones, cambie a un navegador web compatible, como Google Chrome, Mozilla Firefox, Microsoft Edge o Safari.
 iStock_000018808720Large
iStock_000018808720Large Hands placing last piece of a Puzzle
Hands placing last piece of a Puzzle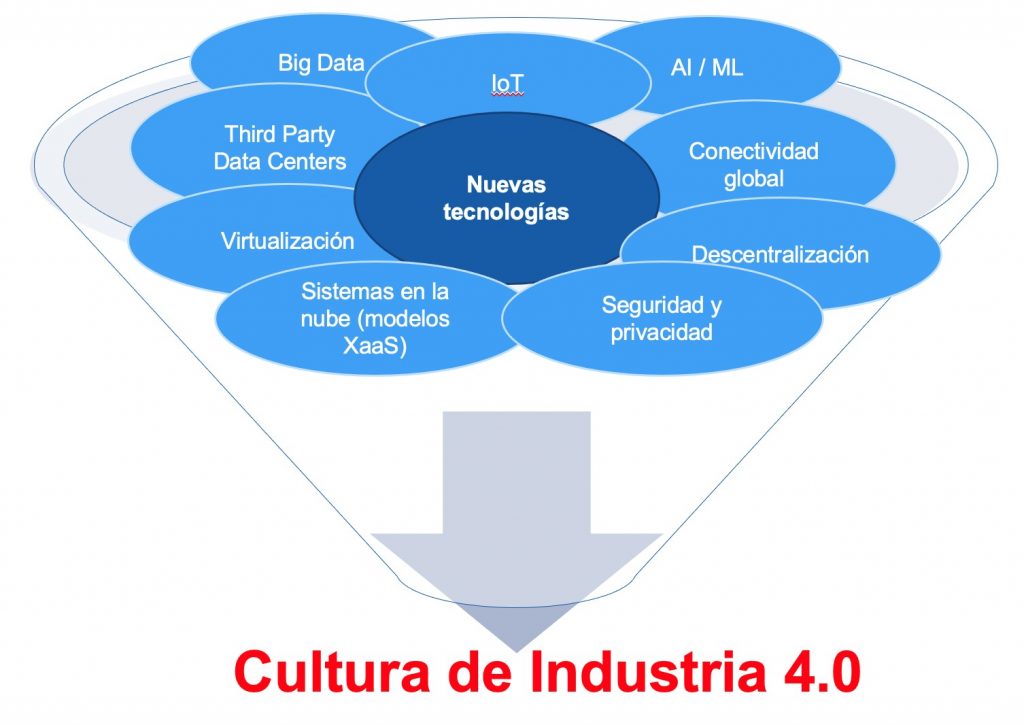
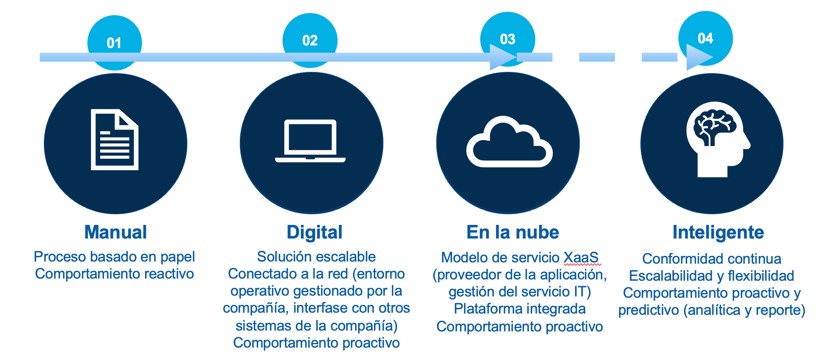
 iStock_000020268060Large
iStock_000020268060Large GMP Qualification and validation
GMP Qualification and validation
 iStock_000016752326Large
iStock_000016752326Large Solutions
Solutions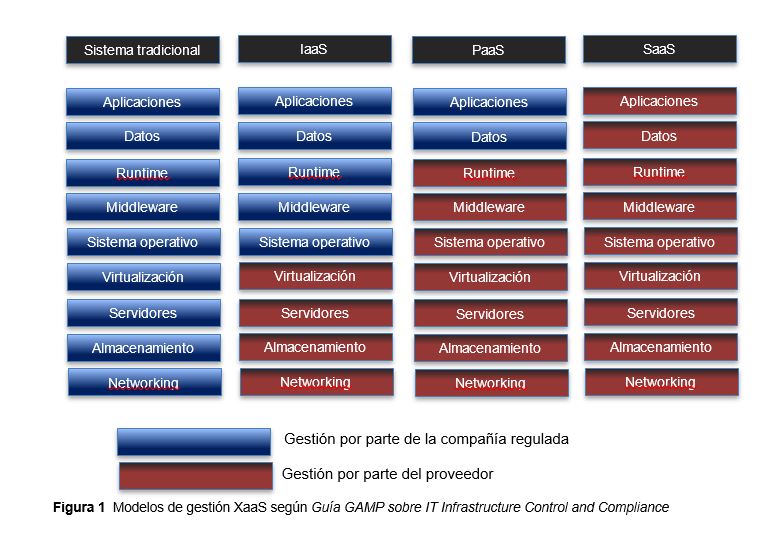
 iStock_000019934130Large
iStock_000019934130Large